

Conpair: concordance and contamination estimator for matched tumor-normal pairs
- PMID: 27354699
- PMCID: PMC5048070
- DOI: 10.1093/bioinformatics/btw389
Conpair: concordance and contamination estimator for matched tumor-normal pairs
Abstract
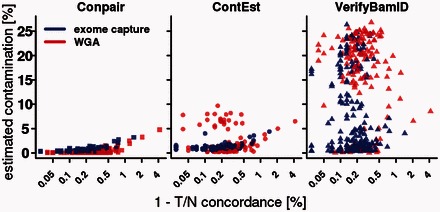
Motivation: Sequencing of matched tumor and normal samples is the standard study design for reliable detection of somatic alterations. However, even very low levels of cross-sample contamination significantly impact calling of somatic mutations, because contaminant germline variants can be incorrectly interpreted as somatic. There are currently no sequence-only based methods that reliably estimate contamination levels in tumor samples, which frequently display copy number changes. As a solution, we developed Conpair, a tool for detection of sample swaps and cross-individual contamination in whole-genome and whole-exome tumor-normal sequencing experiments.
Results: On a ladder of in silico contaminated samples, we demonstrated that Conpair reliably measures contamination levels as low as 0.1%, even in presence of copy number changes. We also estimated contamination levels in glioblastoma WGS and WXS tumor-normal datasets from TCGA and showed that they strongly correlate with tumor-normal concordance, as well as with the number of germline variants called as somatic by several widely-used somatic callers.
Availability and implementation: The method is available at: https://github.com/nygenome/conpair CONTACT: egrabowska@gmail.com or mczody@nygenome.orgSupplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2016. Published by Oxford University Press.
Figures
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
