

AutoSite: an automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms
- PMID: 27354702
- PMCID: PMC5048065
- DOI: 10.1093/bioinformatics/btw367
AutoSite: an automated approach for pseudo-ligands prediction-from ligand-binding sites identification to predicting key ligand atoms
Abstract
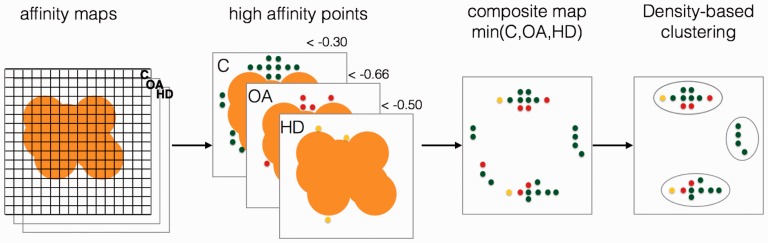
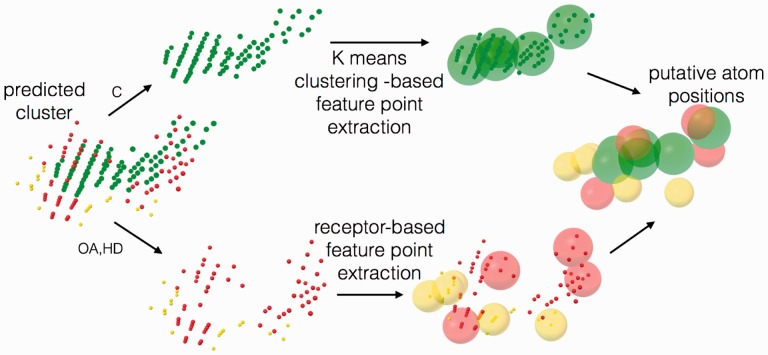
Motivation: The identification of ligand-binding sites from a protein structure facilitates computational drug design and optimization, and protein function assignment. We introduce AutoSite: an efficient software tool for identifying ligand-binding sites and predicting pseudo ligand corresponding to each binding site identified. Binding sites are reported as clusters of 3D points called fills in which every point is labelled as hydrophobic or as hydrogen bond donor or acceptor. From these fills AutoSite derives feature points: a set of putative positions of hydrophobic-, and hydrogen-bond forming ligand atoms.
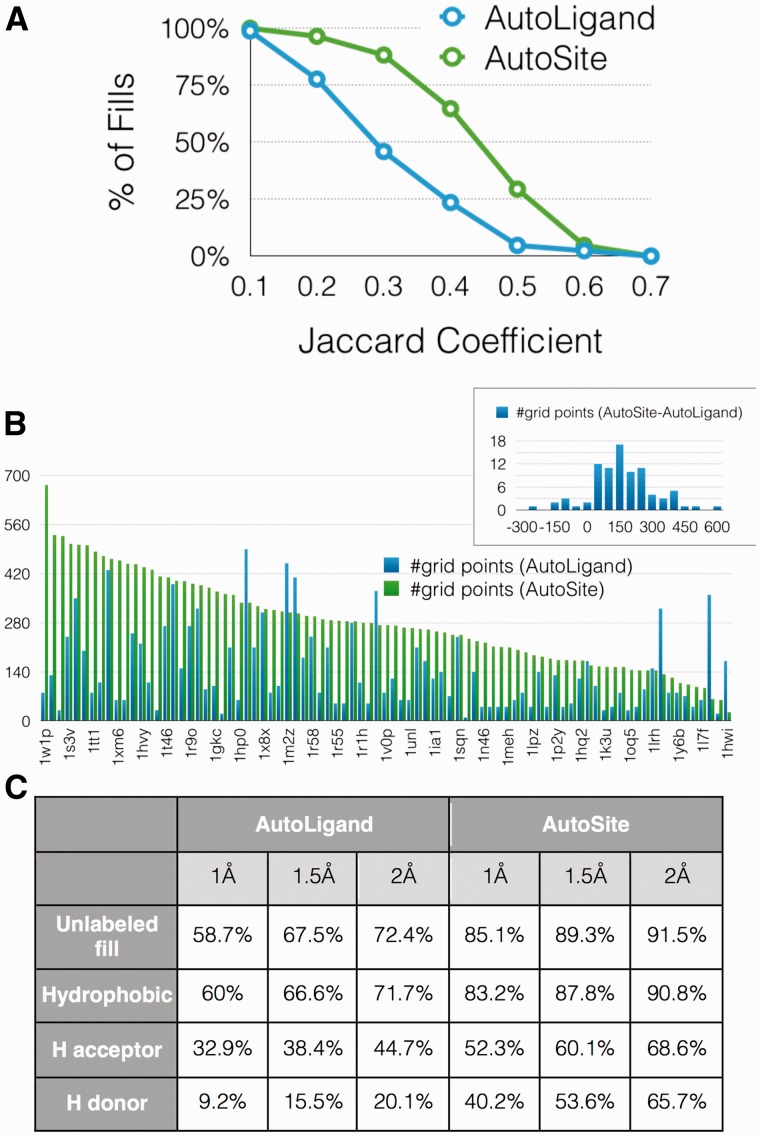
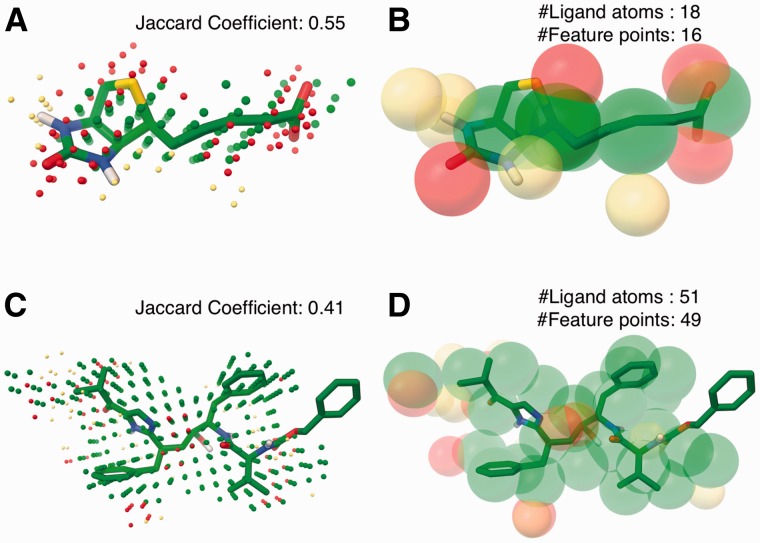
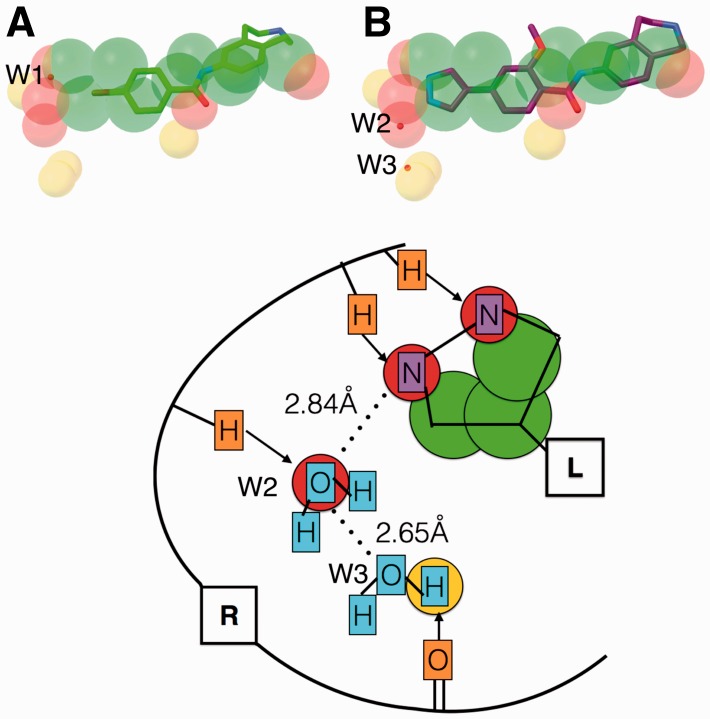
Results: We show that AutoSite identifies ligand-binding sites with higher accuracy than other leading methods, and produces fills that better matches the ligand shape and properties, than the fills obtained with a software program with similar capabilities, AutoLigand In addition, we demonstrate that for the Astex Diverse Set, the feature points identify 79% of hydrophobic ligand atoms, and 81% and 62% of the hydrogen acceptor and donor hydrogen ligand atoms interacting with the receptor, and predict 81.2% of water molecules mediating interactions between ligand and receptor. Finally, we illustrate potential uses of the predicted feature points in the context of lead optimization in drug discovery projects.
Availability and implementation: http://adfr.scripps.edu/AutoDockFR/autosite.html CONTACT: sanner@scripps.eduSupplementary information: Supplementary data are available at Bioinformatics online.
© The Author 2016. Published by Oxford University Press. All rights reserved. For Permissions, please e-mail: journals.permissions@oup.com.
Figures
Similar articles
-
Definition and display of steric, hydrophobic, and hydrogen-bonding properties of ligand binding sites in proteins using Lee and Richards accessible surface: validation of a high-resolution graphical tool for drug design.J Med Chem. 1992 May 15;35(10):1671-84. doi: 10.1021/jm00088a002. J Med Chem. 1992. PMID: 1588550
-
An automated method for predicting the positions of hydrogen-bonding atoms in binding sites.J Comput Aided Mol Des. 1997 May;11(3):229-42. doi: 10.1023/a:1007900527102. J Comput Aided Mol Des. 1997. PMID: 9263850
-
Protein-ligand interfaces are polarized: discovery of a strong trend for intermolecular hydrogen bonds to favor donors on the protein side with implications for predicting and designing ligand complexes.J Comput Aided Mol Des. 2018 Apr;32(4):511-528. doi: 10.1007/s10822-018-0105-2. Epub 2018 Feb 12. J Comput Aided Mol Des. 2018. PMID: 29435780
-
Can We Rely on Computational Predictions To Correctly Identify Ligand Binding Sites on Novel Protein Drug Targets? Assessment of Binding Site Prediction Methods and a Protocol for Validation of Predicted Binding Sites.Cell Biochem Biophys. 2017 Mar;75(1):15-23. doi: 10.1007/s12013-016-0769-y. Epub 2016 Oct 31. Cell Biochem Biophys. 2017. PMID: 27796788 Review.
-
Relibase: design and development of a database for comprehensive analysis of protein-ligand interactions.J Mol Biol. 2003 Feb 14;326(2):607-20. doi: 10.1016/s0022-2836(02)01408-0. J Mol Biol. 2003. PMID: 12559926 Review.
Cited by
-
Rational design of the zonulin inhibitor AT1001 derivatives as potential anti SARS-CoV-2.Eur J Med Chem. 2022 Dec 15;244:114857. doi: 10.1016/j.ejmech.2022.114857. Epub 2022 Oct 19. Eur J Med Chem. 2022. PMID: 36332548 Free PMC article.
-
Allosteric modulation of the CXCR4:CXCL12 axis by targeting receptor nanoclustering via the TMV-TMVI domain.Elife. 2024 Sep 9;13:RP93968. doi: 10.7554/eLife.93968. Elife. 2024. PMID: 39248648 Free PMC article.
-
ProtVar: mapping and contextualizing human missense variation.Nucleic Acids Res. 2024 Jul 5;52(W1):W140-W147. doi: 10.1093/nar/gkae413. Nucleic Acids Res. 2024. PMID: 38769064 Free PMC article.
-
Selective and Effective: Current Progress in Computational Structure-Based Drug Discovery of Targeted Covalent Inhibitors.Trends Pharmacol Sci. 2020 Dec;41(12):1038-1049. doi: 10.1016/j.tips.2020.10.005. Epub 2020 Nov 2. Trends Pharmacol Sci. 2020. PMID: 33153778 Free PMC article. Review.
-
Antiangiogenic potential of phytochemicals from Clerodendrum inerme (L.) Gaertn investigated through in silico and quantum computational methods.Mol Divers. 2025 Feb;29(1):215-239. doi: 10.1007/s11030-024-10846-4. Epub 2024 Apr 28. Mol Divers. 2025. PMID: 38678137
References
-
- An J. et al. (2004) Comprehensive identification of “druggable” protein ligand binding sites. Genome Inform, 15, 31–41. - PubMed
-
- Baroni M. et al. (2007) A common reference framework for analyzing/comparing proteins and ligands. Fingerprints for Ligands and Proteins (FLAP): theory and application. J Chem Inf Model, 47, 279–294. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
