

A Track Record on SHOX: From Basic Research to Complex Models and Therapy
- PMID: 27355317
- PMCID: PMC4971310
- DOI: 10.1210/er.2016-1036
A Track Record on SHOX: From Basic Research to Complex Models and Therapy
Abstract
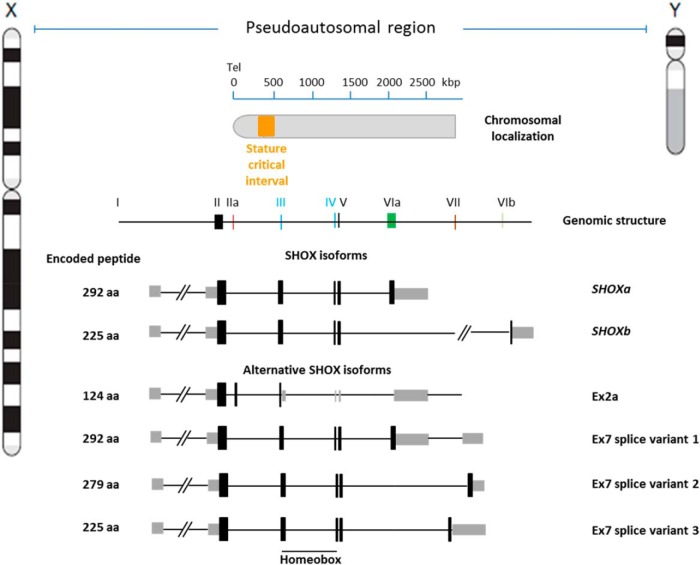
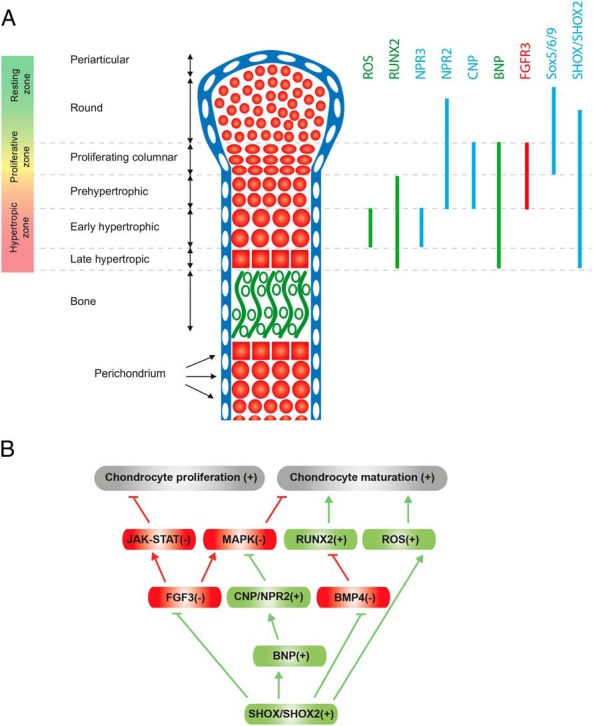
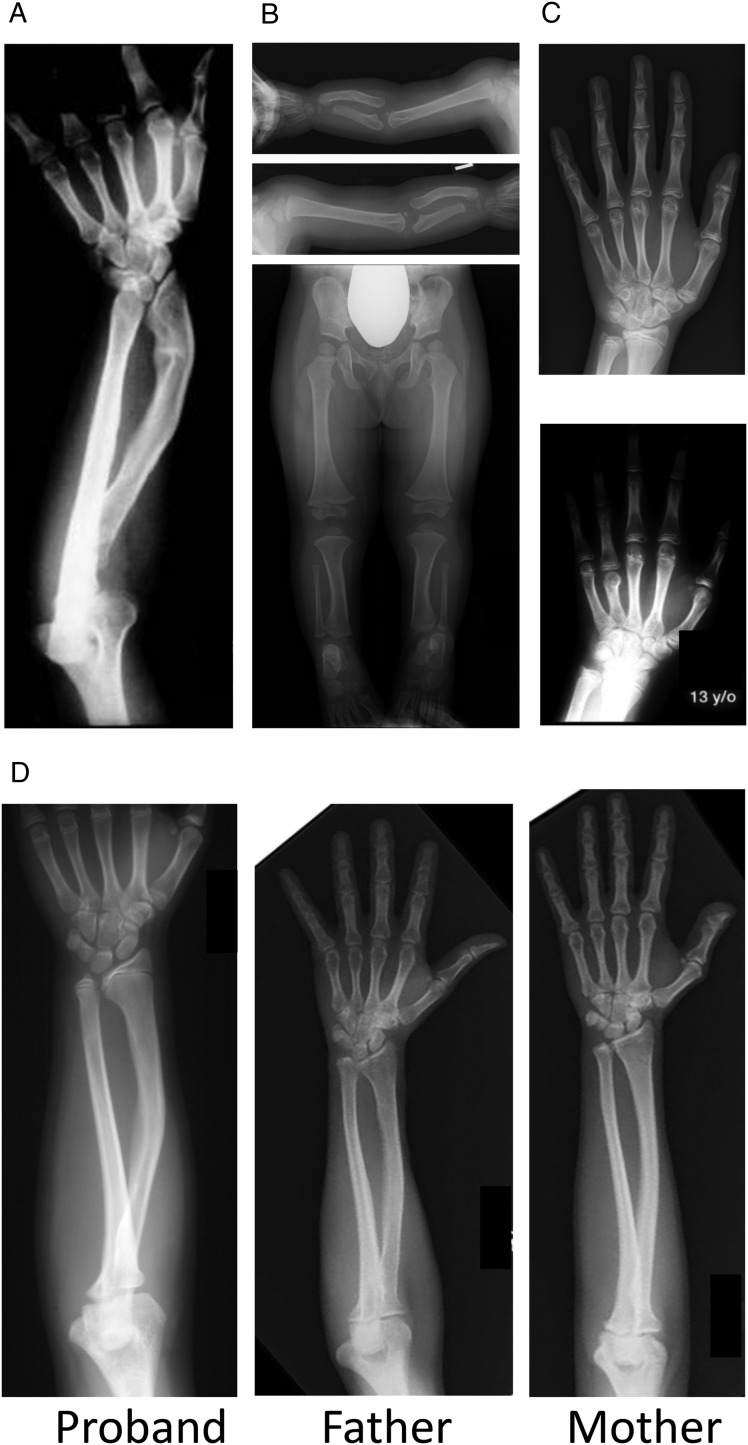
SHOX deficiency is the most frequent genetic growth disorder associated with isolated and syndromic forms of short stature. Caused by mutations in the homeobox gene SHOX, its varied clinical manifestations include isolated short stature, Léri-Weill dyschondrosteosis, and Langer mesomelic dysplasia. In addition, SHOX deficiency contributes to the skeletal features in Turner syndrome. Causative SHOX mutations have allowed downstream pathology to be linked to defined molecular lesions. Expression levels of SHOX are tightly regulated, and almost half of the pathogenic mutations have affected enhancers. Clinical severity of SHOX deficiency varies between genders and ranges from normal stature to profound mesomelic skeletal dysplasia. Treatment options for children with SHOX deficiency are available. Two decades of research support the concept of SHOX as a transcription factor that integrates diverse aspects of bone development, growth plate biology, and apoptosis. Due to its absence in mouse, the animal models of choice have become chicken and zebrafish. These models, therefore, together with micromass cultures and primary cell lines, have been used to address SHOX function. Pathway and network analyses have identified interactors, target genes, and regulators. Here, we summarize recent data and give insight into the critical molecular and cellular functions of SHOX in the etiopathogenesis of short stature and limb development.
Figures




Similar articles
-
SHOX: growth, Léri-Weill and Turner syndromes.Trends Endocrinol Metab. 2000 Aug;11(6):227-30. doi: 10.1016/s1043-2760(00)00262-9. Trends Endocrinol Metab. 2000. PMID: 10878753 Review.
-
Short stature and dysmorphology associated with defects in the SHOX gene.Hormones (Athens). 2006 Apr-Jun;5(2):107-18. doi: 10.14310/horm.2002.11174. Hormones (Athens). 2006. PMID: 16807223 Review.
-
SHOX at a glance: from gene to protein.Arch Physiol Biochem. 2007 Jun;113(3):116-23. doi: 10.1080/13813450701531201. Arch Physiol Biochem. 2007. PMID: 17922307 Review.
-
Combined achondroplasia and short stature homeobox-containing (SHOX) gene deletion in a Danish infant.Eur J Med Genet. 2024 Feb;67:104894. doi: 10.1016/j.ejmg.2023.104894. Epub 2023 Dec 7. Eur J Med Genet. 2024. PMID: 38070826
-
FGFR3 is a target of the homeobox transcription factor SHOX in limb development.Hum Mol Genet. 2011 Apr 15;20(8):1524-35. doi: 10.1093/hmg/ddr030. Epub 2011 Jan 27. Hum Mol Genet. 2011. PMID: 21273290
Cited by
-
A Genetic Approach in the Evaluation of Short Stature.Eurasian J Med. 2022 Dec;54(Suppl1):179-186. doi: 10.5152/eurasianjmed.2022.22171. Eurasian J Med. 2022. PMID: 36655465 Free PMC article.
-
Growth Hormone Treatment for Non-GHD Disorders: Excitement Tempered by Biology.J Clin Endocrinol Metab. 2024 Jan 18;109(2):e442-e454. doi: 10.1210/clinem/dgad417. J Clin Endocrinol Metab. 2024. PMID: 37450564 Free PMC article. Review.
-
Expression levels and DNA methylation profiles of the growth gene SHOX in cartilage tissues and chondrocytes.Sci Rep. 2024 Apr 5;14(1):8069. doi: 10.1038/s41598-024-58530-9. Sci Rep. 2024. PMID: 38580675 Free PMC article.
-
A gene desert required for regulatory control of pleiotropic Shox2 expression and embryonic survival.Nat Commun. 2024 Oct 10;15(1):8793. doi: 10.1038/s41467-024-53009-7. Nat Commun. 2024. PMID: 39389973 Free PMC article.
-
Heterozygous Deletion of the SHOX Gene Enhancer in two Females With Clinical Heterogeneity Associating With Skewed XCI and Escaping XCI.Front Genet. 2019 Nov 6;10:1086. doi: 10.3389/fgene.2019.01086. eCollection 2019. Front Genet. 2019. PMID: 31781162 Free PMC article.
References
-
- Ranke MB. The KIGS aetiology classification system. In: Ranke MB, Gunnarson R, eds. Progress in Growth Hormone Therapy - 5 Years of KIGS. Mannheim, Germany: J&J Verlag; 1994.
-
- Durand C, Rappold GA. Height matters-from monogenic disorders to normal variation. Nat Rev Endocrinol. 2013;9:171–177. - PubMed
-
- Wit JM, Oostdijk W, Losekoot M, van Duyvenvoorde HA, Ruivenkamp CA, Kant SG. Mechanisms in endocrinology: novel genetic causes of short stature. Eur J Endocrinol. 2016;174:R145–R173. - PubMed
-
- Fukami M, Naiki Y, Muroya K, et al. Rare pseudoautosomal copy-number variations involving SHOX and/or its flanking regions in individuals with and without short stature. J Hum Genet. 2015;60:553–556. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
