

Long-read sequencing and de novo assembly of a Chinese genome
- PMID: 27356984
- PMCID: PMC4931320
- DOI: 10.1038/ncomms12065
Long-read sequencing and de novo assembly of a Chinese genome
Abstract
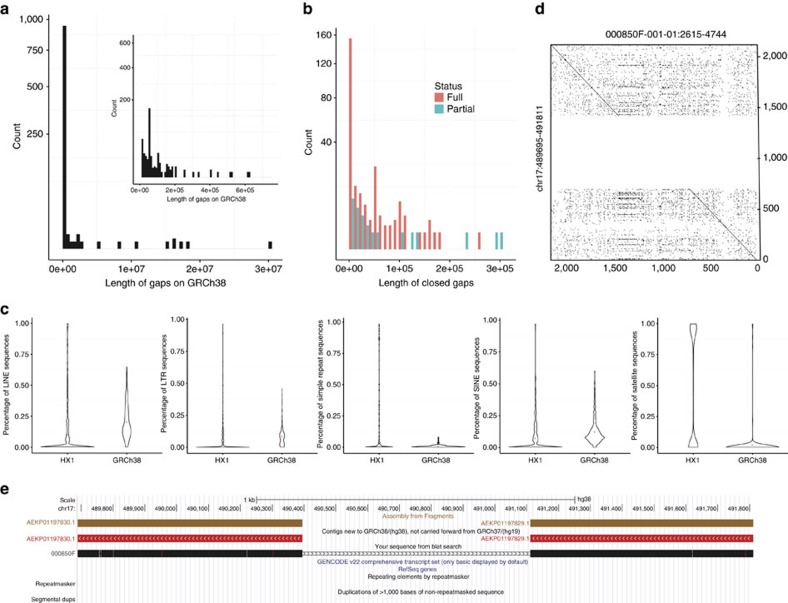
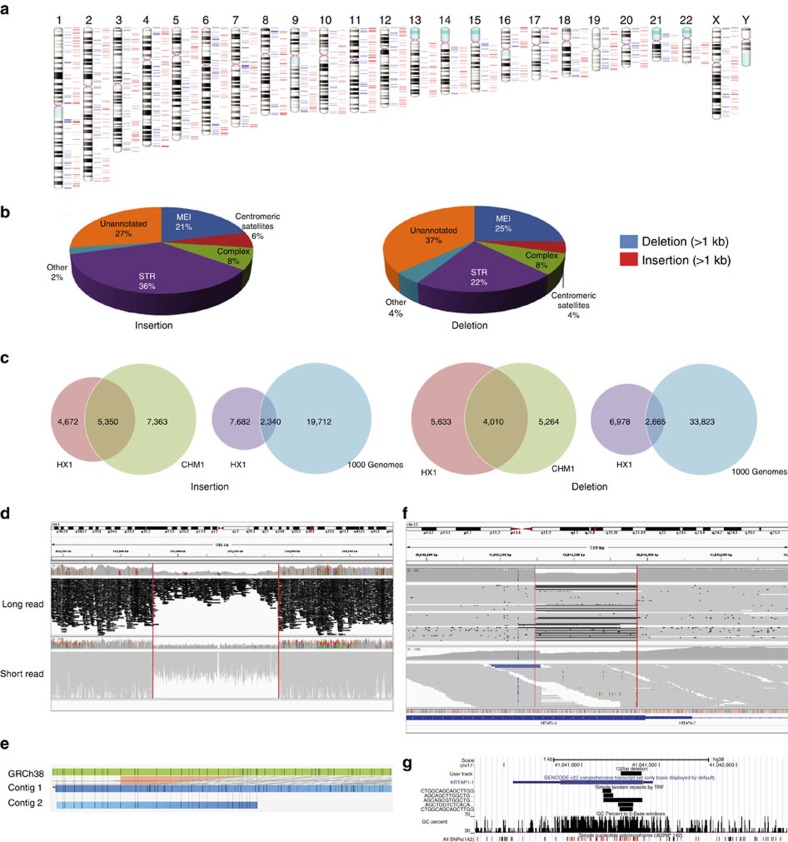
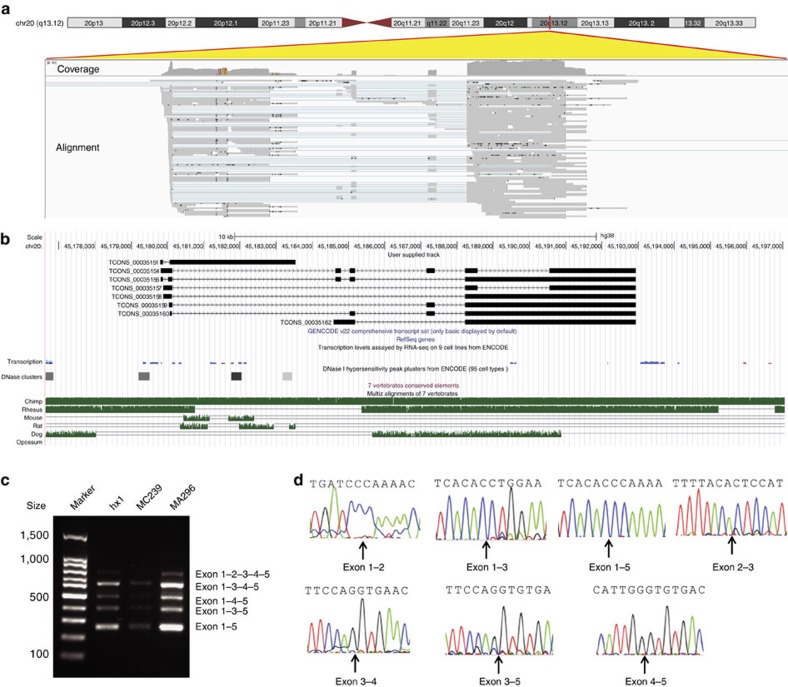
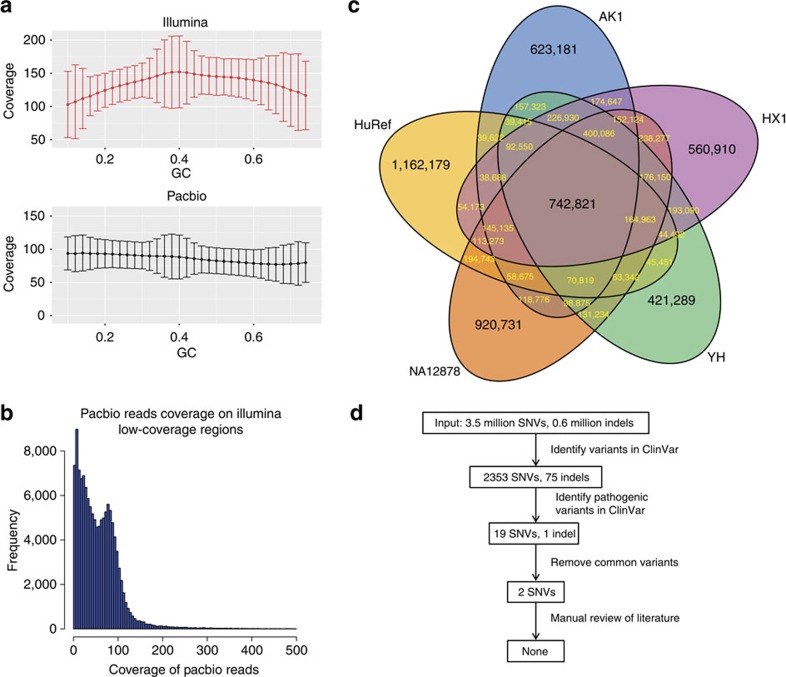
Short-read sequencing has enabled the de novo assembly of several individual human genomes, but with inherent limitations in characterizing repeat elements. Here we sequence a Chinese individual HX1 by single-molecule real-time (SMRT) long-read sequencing, construct a physical map by NanoChannel arrays and generate a de novo assembly of 2.93 Gb (contig N50: 8.3 Mb, scaffold N50: 22.0 Mb, including 39.3 Mb N-bases), together with 206 Mb of alternative haplotypes. The assembly fully or partially fills 274 (28.4%) N-gaps in the reference genome GRCh38. Comparison to GRCh38 reveals 12.8 Mb of HX1-specific sequences, including 4.1 Mb that are not present in previously reported Asian genomes. Furthermore, long-read sequencing of the transcriptome reveals novel spliced genes that are not annotated in GENCODE and are missed by short-read RNA-Seq. Our results imply that improved characterization of genome functional variation may require the use of a range of genomic technologies on diverse human populations.
Conflict of interest statement
J.Hu and D.W. are employees of Nextomics Biosciences. G.J.L. serves on the advisory boards of Omicia, Inc., GenePeeks, Inc. and Good Start Genetics, Inc. K.W. is a board member and shareholder of Tute Genomics, Inc. and Nextomics Biosciences. E.E.E. is on the scientific advisory board of DNAnexus, Inc. and is a consultant for Kunming University of Science and Technology (KUST) as part of the 1000 China Talent Program. The remaining authors declare no competing financial interests.
Figures
References
-
- Cao H. et al. De novo assembly of a haplotype-resolved human genome. Nat. Biotechnol. 33, 617–622 (2015). - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
