

VPAC1 receptor (Vipr1)-deficient mice exhibit ameliorated experimental autoimmune encephalomyelitis, with specific deficits in the effector stage
- PMID: 27357191
- PMCID: PMC4928347
- DOI: 10.1186/s12974-016-0626-3
VPAC1 receptor (Vipr1)-deficient mice exhibit ameliorated experimental autoimmune encephalomyelitis, with specific deficits in the effector stage
Erratum in
-
Erratum to: VPAC1 receptor (Vipr1)-deficient mice exhibit ameliorated experimental autoimmune encephalomyelitis, with specific deficits in the effector stage.J Neuroinflammation. 2017 Aug 4;14(1):157. doi: 10.1186/s12974-017-0927-1. J Neuroinflammation. 2017. PMID: 28778207 Free PMC article. No abstract available.
Abstract
Background: Vasoactive intestinal peptide (VIP) and pituitary adenylyl cyclase-activating polypeptide (PACAP) are two highly homologous neuropeptides. In vitro and ex vivo experiments repeatedly demonstrate that these peptides exert pronounced immunomodulatory (primarily anti-inflammatory) actions which are mediated by common VPAC1 and VPAC2 G protein-coupled receptors. In agreement, we have shown that mice deficient in PACAP ligand or VPAC2 receptors exhibit exacerbated experimental autoimmune encephalomyelitis (EAE). However, we observed that VIP-deficient mice are unexpectedly resistant to EAE, suggesting a requirement for this peptide at some stage of disease development. Here, we investigated the involvement of VPAC1 in the development of EAE using a VPAC1-deficient mouse model.
Methods: EAE was induced in wild-type (WT) and VPAC1 knockout (KO) mice using myelin oligodendrocyte glycoprotein 35-55 (MOG35-55), and clinical scores were assessed continuously over 30 days. Immune responses in the spinal cords were determined by histology, real-time PCR and immunofluorescence, and in the draining lymph nodes by antigen-recall assays. The contribution of VPAC1 expression in the immune system to the development of EAE was evaluated by means of adoptive transfer and bone marrow chimera experiments. In other experiments, VPAC1 receptor analogs were given to WT mice.
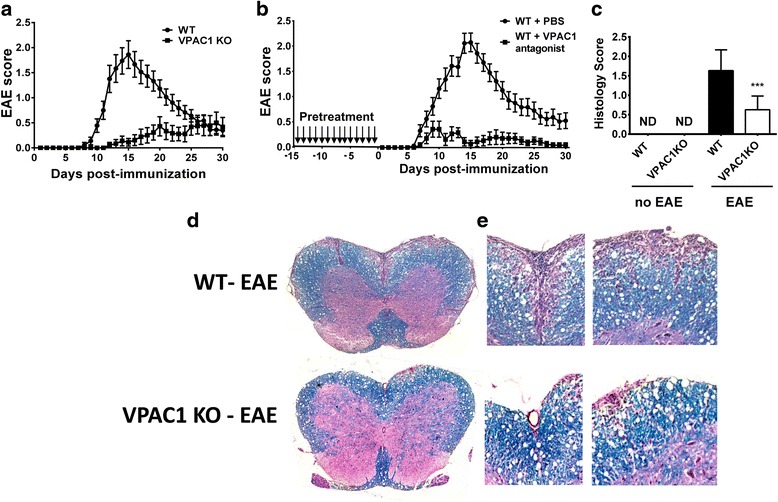
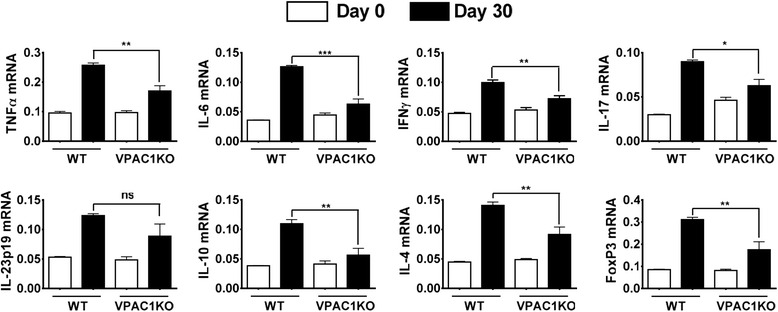
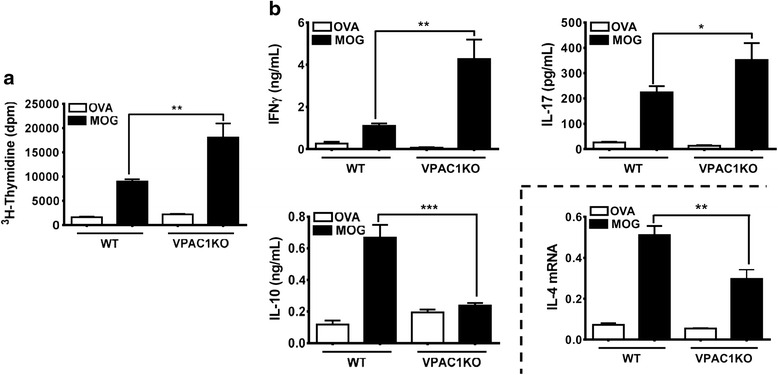
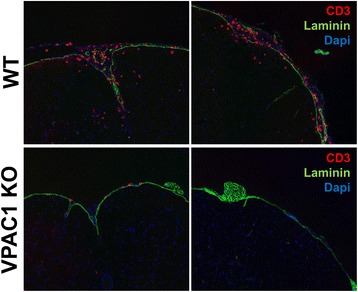
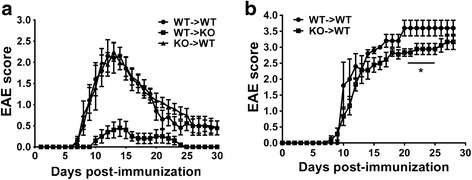
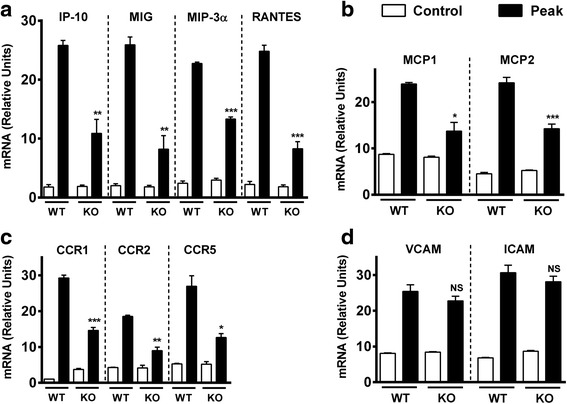
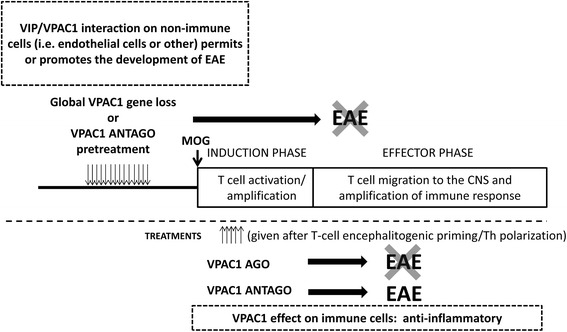
Results: MOG35-55-induced EAE was ameliorated in VPAC1 KO mice compared to WT mice. The EAE-resistant phenotype of VPAC1 KO mice correlated with reduced central nervous system (CNS) histopathology and cytokine expression in the spinal cord. The immunization phase of EAE appeared to be unimpaired because lymph node cells from EAE-induced VPAC1 KO mice stimulated in vitro with MOG exhibited robust proliferative and Th1/Th17 responses. Moreover, lymph node and spleen cells from KO mice were fully capable of inducing EAE upon transfer to WT recipients. In contrast, WT cells from MOG-immunized mice did not transfer the disease when administered to VPAC1 KO recipients, implicating a defect in the effector phase of the disease. Bone marrow chimera studies suggested that the resistance of VPAC1-deficient mice was only minimally dependent on the expression of this receptor in the immunogenic/hematopoietic compartment. Consistent with this, impaired spinal cord inductions of several chemokine mRNAs were observed in VPAC1 KO mice. Finally, treatment of WT mice with the VPAC1 receptor antagonist PG97-269 before, but not after, EAE induction mimicked the clinical phenotype of VPAC1 KO mice.
Conclusions: VPAC1 gene loss impairs the development of EAE in part by preventing an upregulation of CNS chemokines and invasion of inflammatory cells into the CNS. Use of VPAC1 antagonists in WT mice prior to EAE induction also support a critical role for VPAC1 signaling for the development of EAE.
Keywords: Experimental autoimmune encephalomyelitis; Multiple sclerosis; Neuroimmunomodulation; Neuropeptide; VPAC1.
Figures
Similar articles
-
VPAC2 (vasoactive intestinal peptide receptor type 2) receptor deficient mice develop exacerbated experimental autoimmune encephalomyelitis with increased Th1/Th17 and reduced Th2/Treg responses.Brain Behav Immun. 2015 Feb;44:167-175. doi: 10.1016/j.bbi.2014.09.020. Epub 2014 Oct 13. Brain Behav Immun. 2015. PMID: 25305591 Free PMC article.
-
Vasoactive intestinal peptide loss leads to impaired CNS parenchymal T-cell infiltration and resistance to experimental autoimmune encephalomyelitis.Proc Natl Acad Sci U S A. 2010 Nov 9;107(45):19555-60. doi: 10.1073/pnas.1007622107. Epub 2010 Oct 26. Proc Natl Acad Sci U S A. 2010. PMID: 20978211 Free PMC article.
-
Rescue from acute neuroinflammation by pharmacological chemokine-mediated deviation of leukocytes.J Neuroinflammation. 2012 Oct 25;9:243. doi: 10.1186/1742-2094-9-243. J Neuroinflammation. 2012. PMID: 23095573 Free PMC article.
-
Gene therapy in autoimmune, demyelinating disease of the central nervous system.Gene Ther. 2003 May;10(10):844-53. doi: 10.1038/sj.gt.3302025. Gene Ther. 2003. PMID: 12732870 Review.
-
Studies in the modulation of experimental autoimmune encephalomyelitis.J Neuroimmune Pharmacol. 2010 Jun;5(2):168-75. doi: 10.1007/s11481-010-9215-x. Epub 2010 Apr 17. J Neuroimmune Pharmacol. 2010. PMID: 20401539 Free PMC article. Review.
Cited by
-
Multiple Sclerosis: Inflammatory and Neuroglial Aspects.Curr Issues Mol Biol. 2023 Feb 8;45(2):1443-1470. doi: 10.3390/cimb45020094. Curr Issues Mol Biol. 2023. PMID: 36826039 Free PMC article. Review.
-
Immunomodulatory Roles of PACAP and VIP: Lessons from Knockout Mice.J Mol Neurosci. 2018 Sep;66(1):102-113. doi: 10.1007/s12031-018-1150-y. Epub 2018 Aug 13. J Mol Neurosci. 2018. PMID: 30105629 Review.
-
PACAP/PAC1 Regulation of Inflammation via Catecholaminergic Neurons in a Model of Multiple Sclerosis.J Mol Neurosci. 2019 Jul;68(3):439-451. doi: 10.1007/s12031-018-1137-8. Epub 2018 Jul 30. J Mol Neurosci. 2019. PMID: 30058008 Free PMC article.
-
A Clinical Approach for the Use of VIP Axis in Inflammatory and Autoimmune Diseases.Int J Mol Sci. 2019 Dec 20;21(1):65. doi: 10.3390/ijms21010065. Int J Mol Sci. 2019. PMID: 31861827 Free PMC article. Review.
-
Targeting vasoactive intestinal peptide-mediated signaling enhances response to immune checkpoint therapy in pancreatic ductal adenocarcinoma.Nat Commun. 2022 Oct 27;13(1):6418. doi: 10.1038/s41467-022-34242-4. Nat Commun. 2022. PMID: 36302761 Free PMC article.
References
-
- Fahrenkrug J. VIP and PACAP. Results Probl Cell Differ. 2010;50:221–234. - PubMed
-
- Harmar AJ, Fahrenkrug J, Gozes I, Laburthe M, May V, Pisegna JR, Vaudry D, Vaudry H, Waschek JA, Said SI. Pharmacology and functions of receptors for vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide: IUPHAR review 1. Br J Pharmacol. 2012;166:4–17. doi: 10.1111/j.1476-5381.2012.01871.x. - DOI - PMC - PubMed
-
- Delgado M, Munoz-Elias EJ, Kan Y, Gozes I, Fridkin M, Brenneman DE, Gomariz RP, Ganea D. Vasoactive intestinal peptide and pituitary adenylate cyclase-activating polypeptide inhibit tumor necrosis factor alpha transcriptional activation by regulating nuclear factor-kB and cAMP response element-binding protein/c-Jun. J Biol Chem. 1998;273:31427–31436. doi: 10.1074/jbc.273.47.31427. - DOI - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
