

Algorithmic methods to infer the evolutionary trajectories in cancer progression
- PMID: 27357673
- PMCID: PMC4948322
- DOI: 10.1073/pnas.1520213113
Algorithmic methods to infer the evolutionary trajectories in cancer progression
Abstract
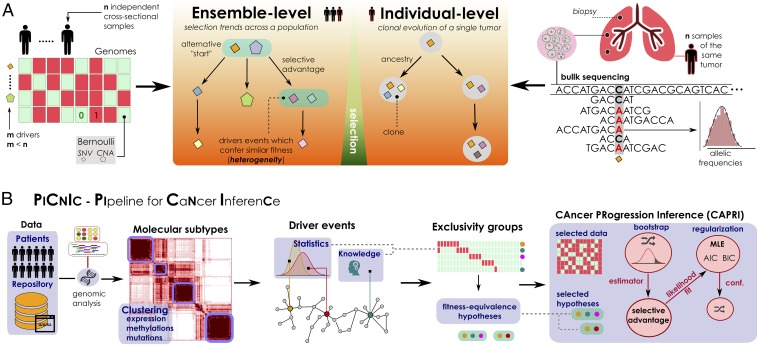
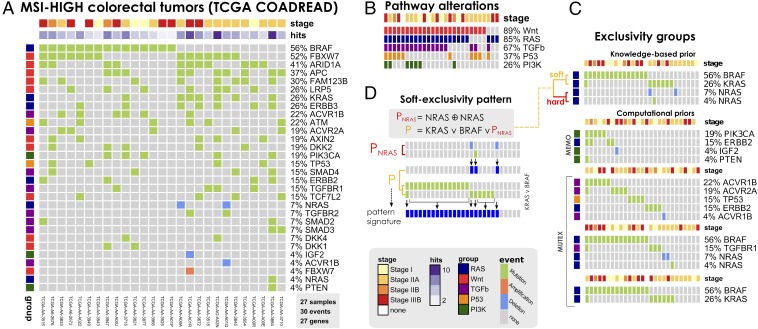
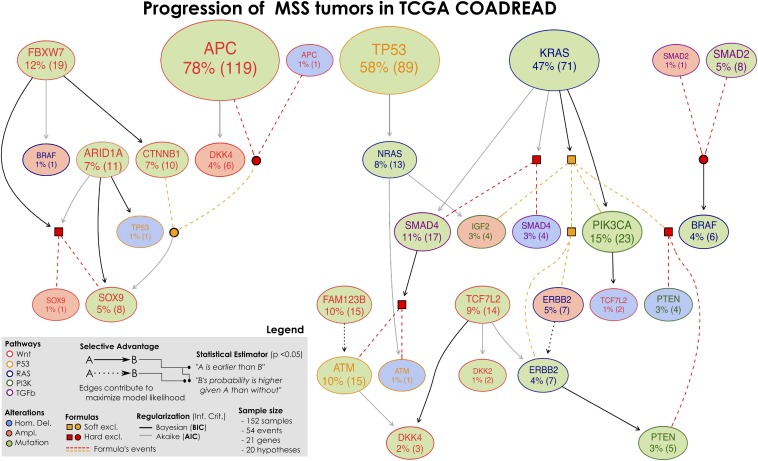
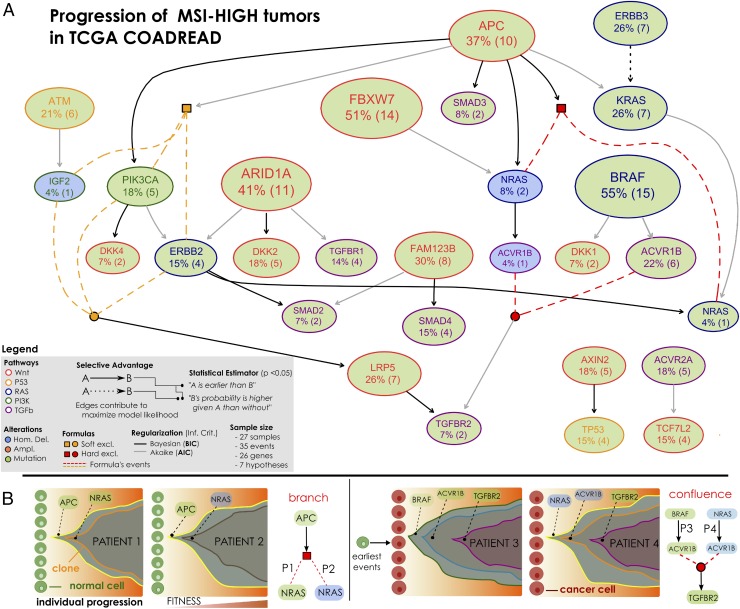
The genomic evolution inherent to cancer relates directly to a renewed focus on the voluminous next-generation sequencing data and machine learning for the inference of explanatory models of how the (epi)genomic events are choreographed in cancer initiation and development. However, despite the increasing availability of multiple additional -omics data, this quest has been frustrated by various theoretical and technical hurdles, mostly stemming from the dramatic heterogeneity of the disease. In this paper, we build on our recent work on the "selective advantage" relation among driver mutations in cancer progression and investigate its applicability to the modeling problem at the population level. Here, we introduce PiCnIc (Pipeline for Cancer Inference), a versatile, modular, and customizable pipeline to extract ensemble-level progression models from cross-sectional sequenced cancer genomes. The pipeline has many translational implications because it combines state-of-the-art techniques for sample stratification, driver selection, identification of fitness-equivalent exclusive alterations, and progression model inference. We demonstrate PiCnIc's ability to reproduce much of the current knowledge on colorectal cancer progression as well as to suggest novel experimentally verifiable hypotheses.
Keywords: Bayesian structural inference; cancer evolution; causality; next generation sequencing; selective advantage.
Conflict of interest statement
The authors declare no conflict of interest.
Figures

References
-
- Nowell PC. The clonal evolution of tumor cell populations. Science. 1976;194(4260):23–28. - PubMed
-
- Fidler IJ. Tumor heterogeneity and the biology of cancer invasion and metastasis. Cancer Res. 1978;38(9):2651–2660. - PubMed
-
- Dexter DL, et al. Heterogeneity of tumor cells from a single mouse mammary tumor. Cancer Res. 1978;38(10):3174–3181. - PubMed
-
- Merlo LM, Pepper JW, Reid BJ, Maley CC. Cancer as an evolutionary and ecological process. Nat Rev Cancer. 2006;6(12):924–935. - PubMed
-
- Hanahan D, Weinberg RA. The hallmarks of cancer. Cell. 2000;100(1):57–70. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
