

Reference-free deconvolution of DNA methylation data and mediation by cell composition effects
- PMID: 27358049
- PMCID: PMC4928286
- DOI: 10.1186/s12859-016-1140-4
Reference-free deconvolution of DNA methylation data and mediation by cell composition effects
Abstract
Background: Recent interest in reference-free deconvolution of DNA methylation data has led to several supervised methods, but these methods do not easily permit the interpretation of underlying cell types.
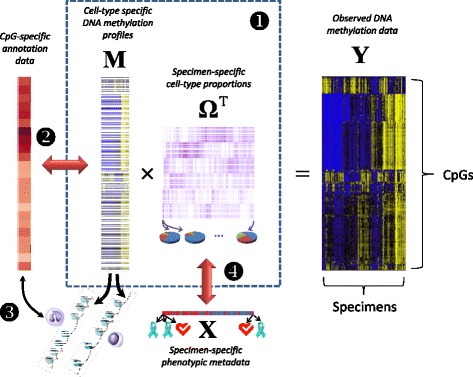
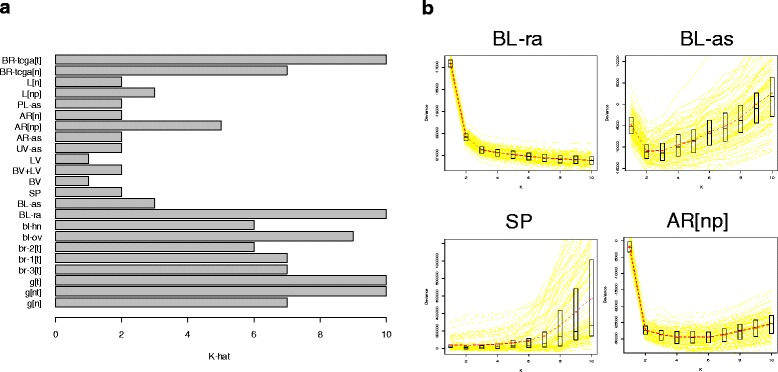
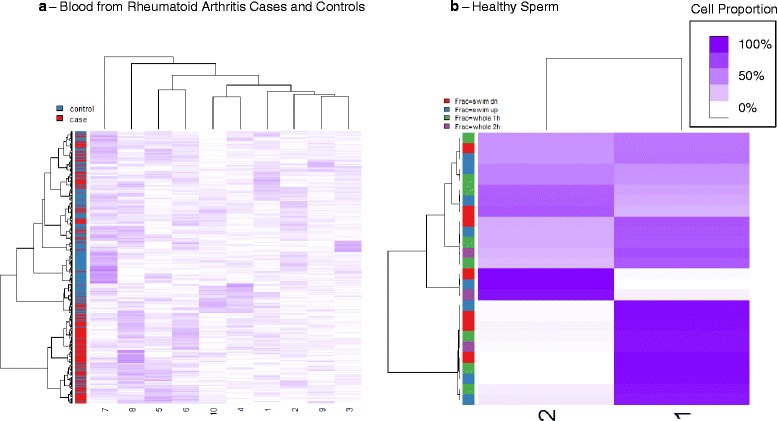
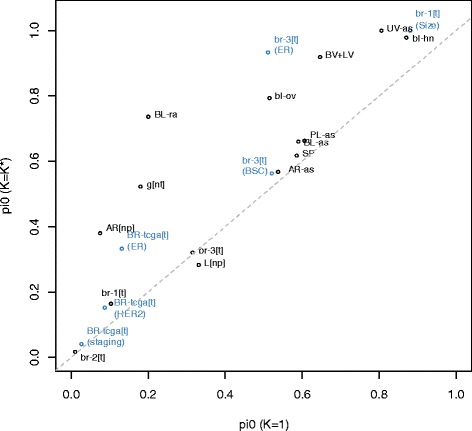
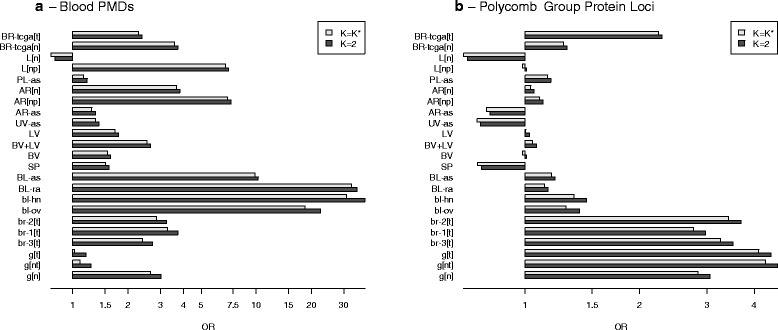
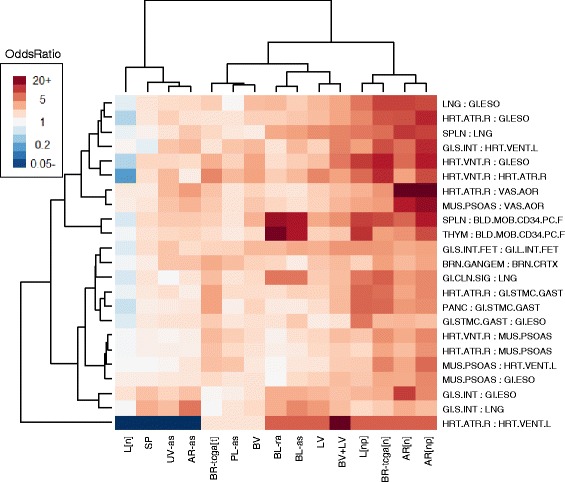
Results: We propose a simple method for reference-free deconvolution that provides both proportions of putative cell types defined by their underlying methylomes, the number of these constituent cell types, as well as a method for evaluating the extent to which the underlying methylomes reflect specific types of cells. We demonstrate these methods in an analysis of 23 Infinium data sets from 13 distinct data collection efforts; these empirical evaluations show that our algorithm can reasonably estimate the number of constituent types, return cell proportion estimates that demonstrate anticipated associations with underlying phenotypic data; and methylomes that reflect the underlying biology of constituent cell types.
Conclusions: Our methodology permits an explicit quantitation of the mediation of phenotypic associations with DNA methylation by cell composition effects. Although more work is needed to investigate functional information related to estimated methylomes, our proposed method provides a novel and useful foundation for conducting DNA methylation studies on heterogeneous tissues lacking reference data.
Keywords: DNA methylation; Deconvolution; Epigenetics; Non-negative matrix factorization.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
