

De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy
- PMID: 27358180
- PMCID: PMC5264224
- DOI: 10.1136/jmedgenet-2016-103909
De novo mutations of KIAA2022 in females cause intellectual disability and intractable epilepsy
Abstract
Background: Mutations in the KIAA2022 gene have been reported in male patients with X-linked intellectual disability, and related female carriers were unaffected. Here, we report 14 female patients who carry a heterozygous de novo KIAA2022 mutation and share a phenotype characterised by intellectual disability and epilepsy.
Methods: Reported females were selected for genetic testing because of substantial developmental problems and/or epilepsy. X-inactivation and expression studies were performed when possible.
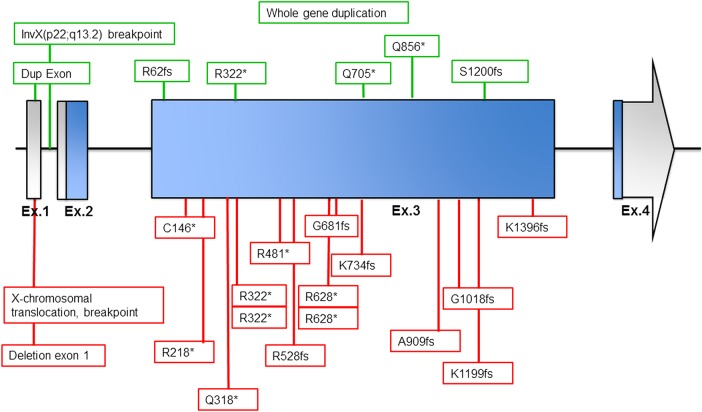
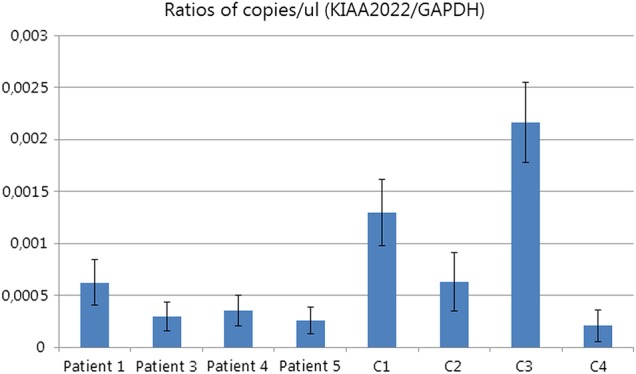
Results: All mutations were predicted to result in a frameshift or premature stop. 12 out of 14 patients had intractable epilepsy with myoclonic and/or absence seizures, and generalised in 11. Thirteen patients had mild to severe intellectual disability. This female phenotype partially overlaps with the reported male phenotype which consists of more severe intellectual disability, microcephaly, growth retardation, facial dysmorphisms and, less frequently, epilepsy. One female patient showed completely skewed X-inactivation, complete absence of RNA expression in blood and a phenotype similar to male patients. In the six other tested patients, X-inactivation was random, confirmed by a non-significant twofold to threefold decrease of RNA expression in blood, consistent with the expected mosaicism between cells expressing mutant or normal KIAA2022 alleles.
Conclusions: Heterozygous loss of KIAA2022 expression is a cause of intellectual disability in females. Compared with its hemizygous male counterpart, the heterozygous female disease has less severe intellectual disability, but is more often associated with a severe and intractable myoclonic epilepsy.
Keywords: KIAA2022; Clinical genetics; Epilepsy and seizures; X-linked.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Conflict of interest statement
KLH and ST are employed by and receive a salary from Ambry Genetics. BAM was supported by Genome Canada and the Ontario Brain Institute. BM, SB and SS are funded by the Epilepsy Society and Wellcome Trust. Part of this work was undertaken at University College London Hospitals, which received a proportion of funding from the NIHR Biomedical Research Centres funding scheme.
Figures
References
-
- Van Maldergem L, Hou Q, Kalscheuer VM, Rio M, Doco-Fenzy M, Medeira A, de Brouwer APM, Cabrol C, Haas SA, Cacciagli P, Moutton S, Landais E, Motte J, Colleaux L, Bonnet C, Villard L, Dupont J, Man H-Y. Loss of function of KIAA2022 causes mild to severe intellectual disability with an autism spectrum disorder and impairs neurite outgrowth. Hum Mol Genet 2013;22:3306–14. 10.1093/hmg/ddt187 - DOI - PMC - PubMed
-
- Soden SE, Saunders CJ, Willig LK, Farrow EG, Smith LD, Petrikin JE, LePichon J-B, Miller NA, Thiffault I, Dinwiddie DL, Twist G, Noll A, Heese BA, Zellmer L, Atherton AM, Abdelmoity AT, Safina N, Nyp SS, Zuccarelli B, Larson IA, Modrcin A, Herd S, Creed M, Ye Z, Yuan X, Brodsky RA, Kingsmore SF. Effectiveness of exome and genome sequencing guided by acuity of illness for diagnosis of neurodevelopmental disorders. Sci Transl Med 2014;6:265ra168 10.1126/scitranslmed.3010076 - DOI - PMC - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases