

Liver transplantation and the management of progressive familial intrahepatic cholestasis in children
- PMID: 27358773
- PMCID: PMC4919732
- DOI: 10.5500/wjt.v6.i2.278
Liver transplantation and the management of progressive familial intrahepatic cholestasis in children
Abstract
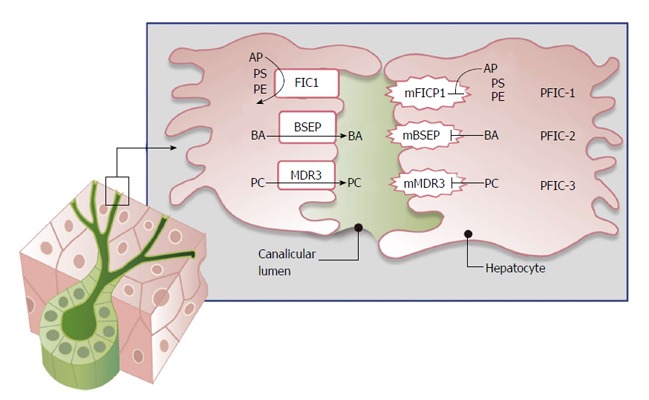
Progressive familial intrahepatic cholestasis (PFIC) is a constellation of inherited disorders that result in the impairment of bile flow through the liver that predominantly affects children. The accumulation of bile results in progressive liver damage, and if left untreated leads to end stage liver disease and death. Patients often present with worsening jaundice and pruritis within the first few years of life. Many of these patients will progress to end stage liver disease and require liver transplantation. The role and timing of liver transplantation still remains debated especially in the management of PFIC1. In those patients who are appropriately selected, liver transplantation offers an excellent survival benefit. Appropriate timing and selection of patients for liver transplantation will be discussed, and the short and long term management of patients post liver transplantation will also be described.
Keywords: Bile salt excretion protein; Cholestasis; Familial intrahepatic cholestasis protein 1; Multidrug resistance protein 3; Pediatric jaundice; Pediatric liver transplant; Progressive familial intrahepatic cholestasis.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
