

Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come
- PMID: 27363439
- PMCID: PMC4931871
- DOI: 10.1124/pr.115.012070
Lipid-Based Drug Delivery Systems in Cancer Therapy: What Is Available and What Is Yet to Come
Abstract
Cancer is a leading cause of death in many countries around the world. However, the efficacy of current standard treatments for a variety of cancers is suboptimal. First, most cancer treatments lack specificity, meaning that these treatments affect both cancer cells and their normal counterparts. Second, many anticancer agents are highly toxic, and thus, limit their use in treatment. Third, a number of cytotoxic chemotherapeutics are highly hydrophobic, which limits their utility in cancer therapy. Finally, many chemotherapeutic agents exhibit short half-lives that curtail their efficacy. As a result of these deficiencies, many current treatments lead to side effects, noncompliance, and patient inconvenience due to difficulties in administration. However, the application of nanotechnology has led to the development of effective nanosized drug delivery systems known commonly as nanoparticles. Among these delivery systems, lipid-based nanoparticles, particularly liposomes, have shown to be quite effective at exhibiting the ability to: 1) improve the selectivity of cancer chemotherapeutic agents; 2) lower the cytotoxicity of anticancer drugs to normal tissues, and thus, reduce their toxic side effects; 3) increase the solubility of hydrophobic drugs; and 4) offer a prolonged and controlled release of agents. This review will discuss the current state of lipid-based nanoparticle research, including the development of liposomes for cancer therapy, different strategies for tumor targeting, liposomal formulation of various anticancer drugs that are commercially available, recent progress in liposome technology for the treatment of cancer, and the next generation of lipid-based nanoparticles.
Copyright © 2016 by The American Society for Pharmacology and Experimental Therapeutics.
Figures











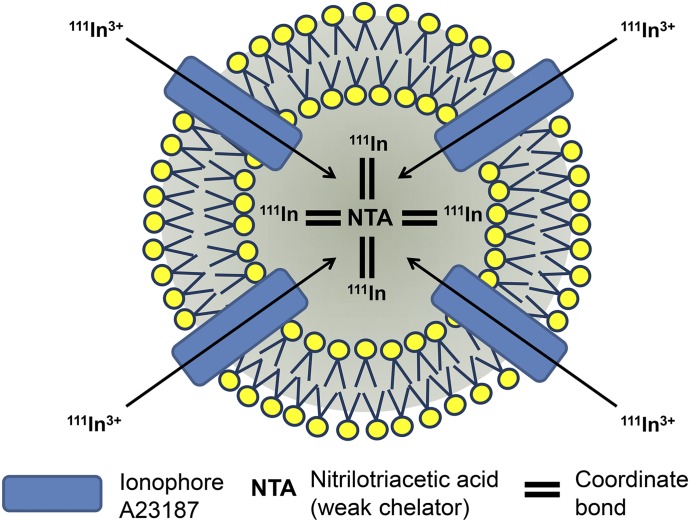


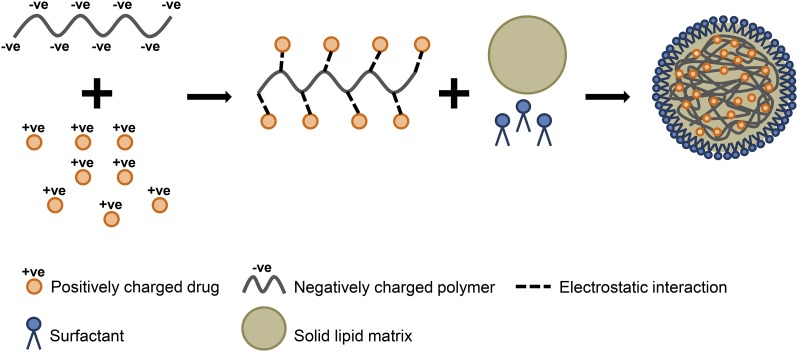



References
-
- Abid MR, Yi X, Yano K, Shih S-C, Aird WC. (2006) Vascular endocan is preferentially expressed in tumor endothelium. Microvasc Res 72:136–145. - PubMed
-
- Abuchowski A, McCoy JR, Palczuk NC, van Es T, Davis FF. (1977) Effect of covalent attachment of polyethylene glycol on immunogenicity and circulating life of bovine liver catalase. J Biol Chem 252:3582–3586. - PubMed
-
- Ahmed SE, Martins AM, Husseini GA. (2015) The use of ultrasound to release chemotherapeutic drugs from micelles and liposomes. J Drug Target 23:16–42. - PubMed
-
- Alkan-Onyuksel H, Ramakrishnan S, Chai H-B, Pezzuto JM. (1994) A mixed micellar formulation suitable for the parenteral administration of taxol. Pharm Res 11:206–212. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
