

Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer
- PMID: 27372738
- PMCID: PMC4967013
- DOI: 10.1016/j.cell.2016.05.069
Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer
Erratum in
-
Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer.Cell. 2025 Nov 26;188(24):7016. doi: 10.1016/j.cell.2025.10.043. Epub 2025 Nov 4. Cell. 2025. PMID: 41187752 No abstract available.
Abstract
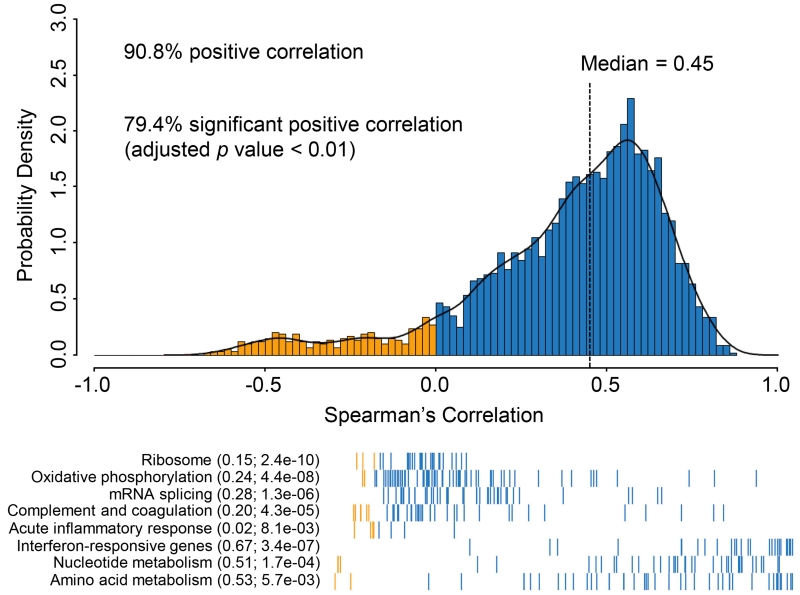
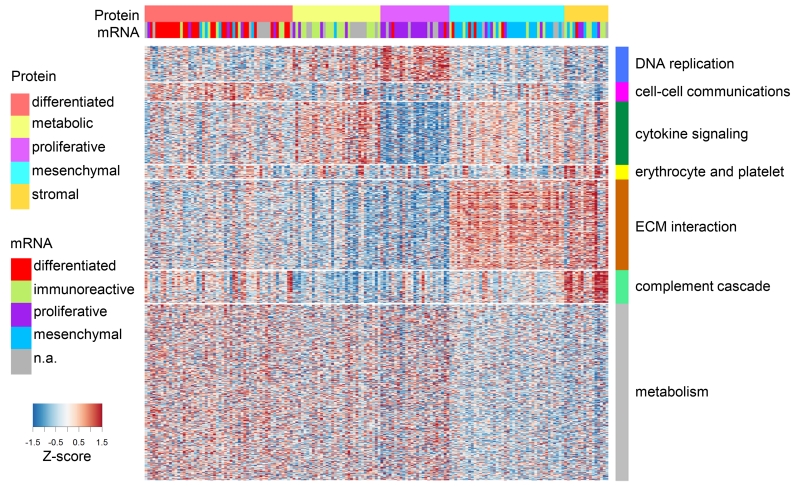
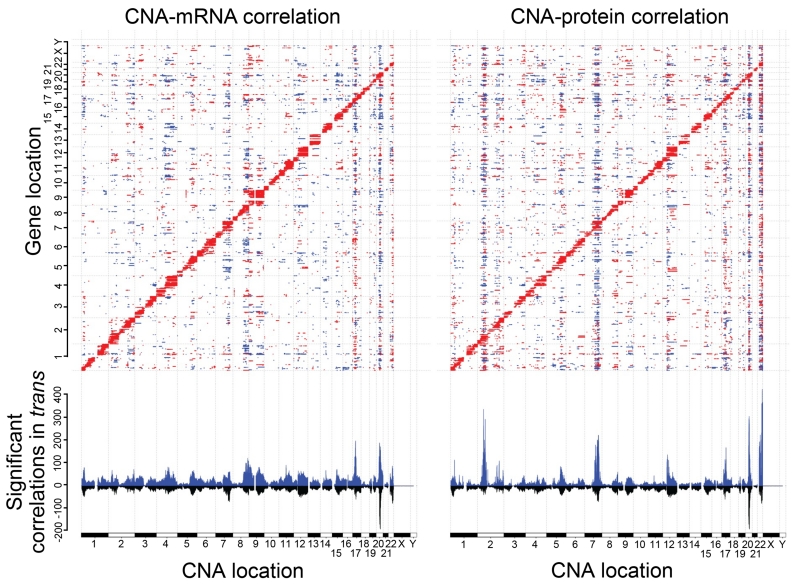
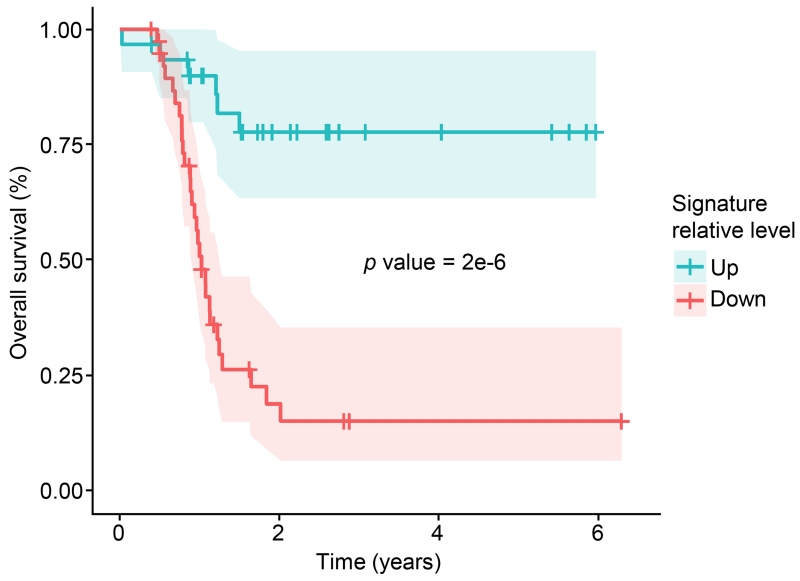
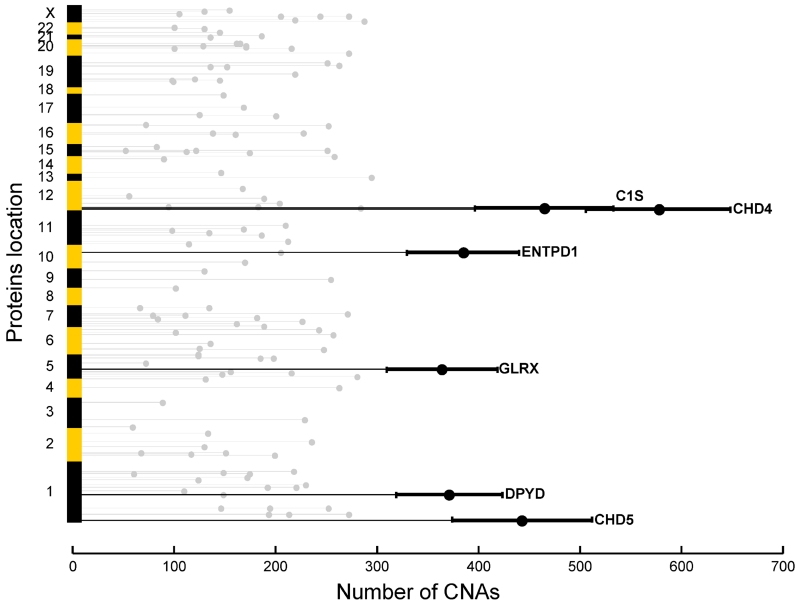
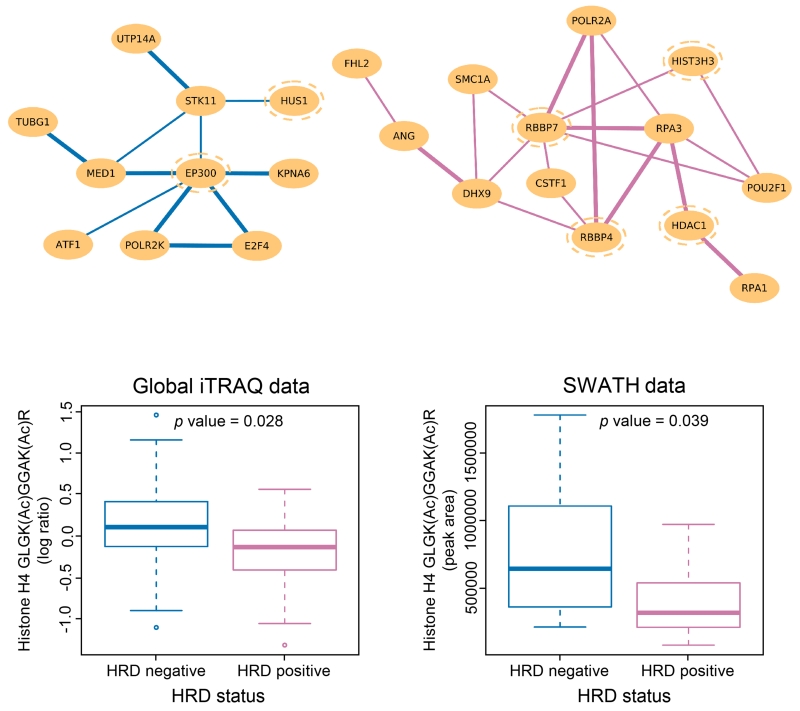
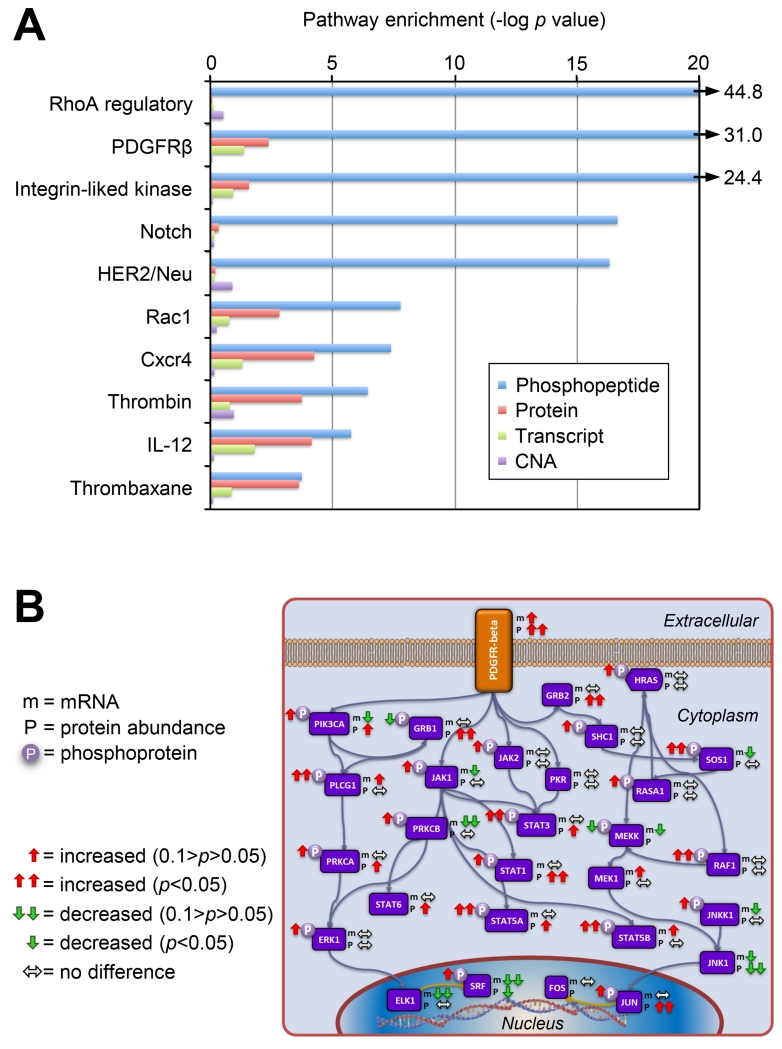
To provide a detailed analysis of the molecular components and underlying mechanisms associated with ovarian cancer, we performed a comprehensive mass-spectrometry-based proteomic characterization of 174 ovarian tumors previously analyzed by The Cancer Genome Atlas (TCGA), of which 169 were high-grade serous carcinomas (HGSCs). Integrating our proteomic measurements with the genomic data yielded a number of insights into disease, such as how different copy-number alternations influence the proteome, the proteins associated with chromosomal instability, the sets of signaling pathways that diverse genome rearrangements converge on, and the ones most associated with short overall survival. Specific protein acetylations associated with homologous recombination deficiency suggest a potential means for stratifying patients for therapy. In addition to providing a valuable resource, these findings provide a view of how the somatic genome drives the cancer proteome and associations between protein and post-translational modification levels and clinical outcomes in HGSC. VIDEO ABSTRACT.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
References
-
- Cai RL, Yan-Neale Y, Cueto MA, Xu H, Cohen D. HDAC1, a histone deacetylase, forms a complex with Hus1 and Rad9, two G2/M checkpoint Rad proteins. J. Biol. Chem. 2000;275:27909–27916. - PubMed
-
- Collins BC, Gillet LC, Rosenberger G, Rost HL, Vichalkovski A, Gstaiger M, Aebersold R. Quantifying protein interaction dynamics by SWATH mass spectrometry: application to the 14-3-3 system. Nat. Methods. 2013;10:1246–1253. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
