

Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome
- PMID: 27374344
- PMCID: PMC5105181
- DOI: 10.1165/rcmb.2016-0008OC
Functional Characterization of ATP-Binding Cassette Transporter A3 Mutations from Infants with Respiratory Distress Syndrome
Abstract
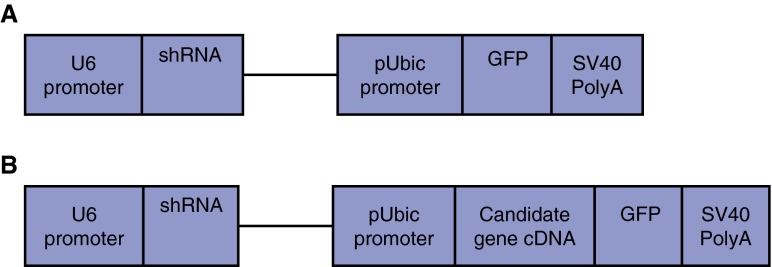
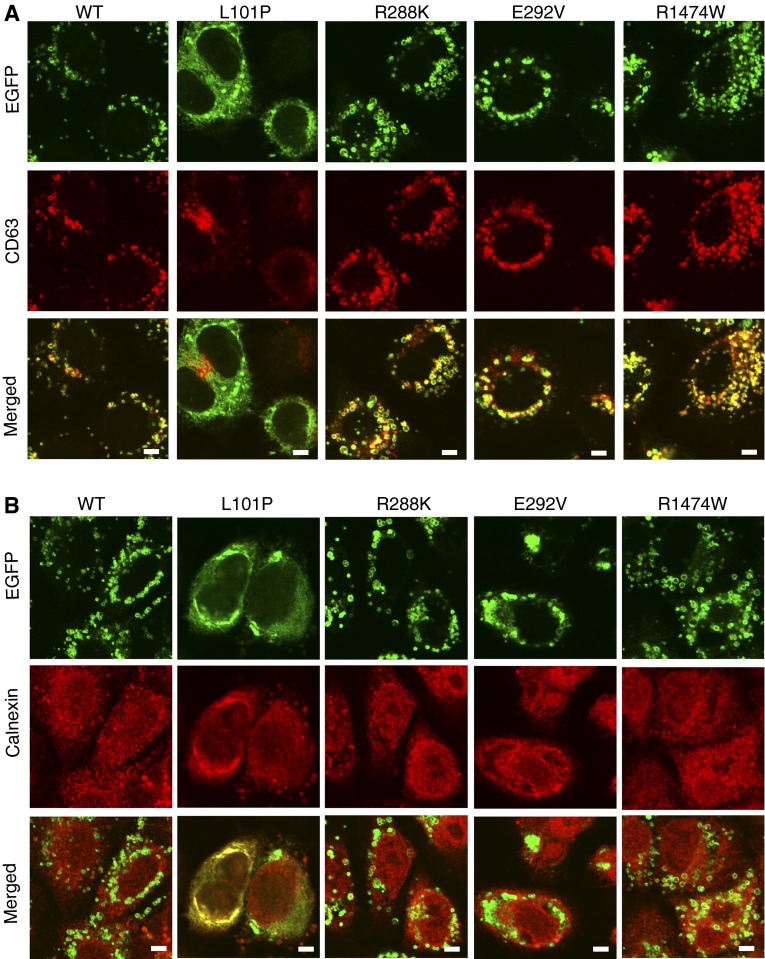
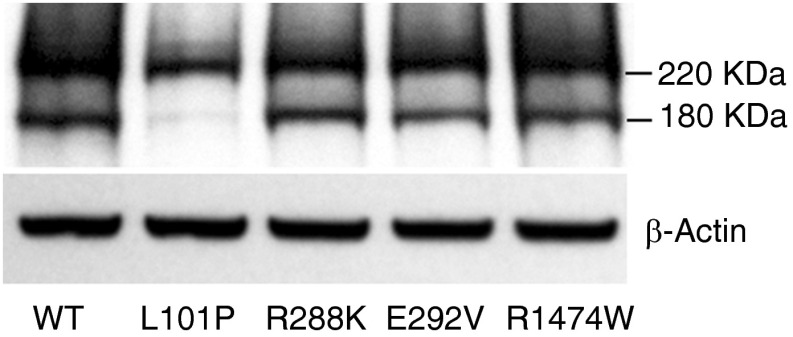
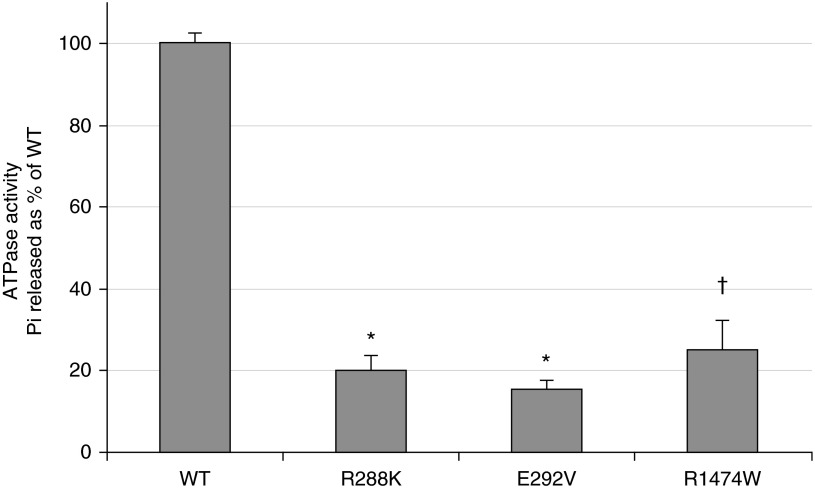
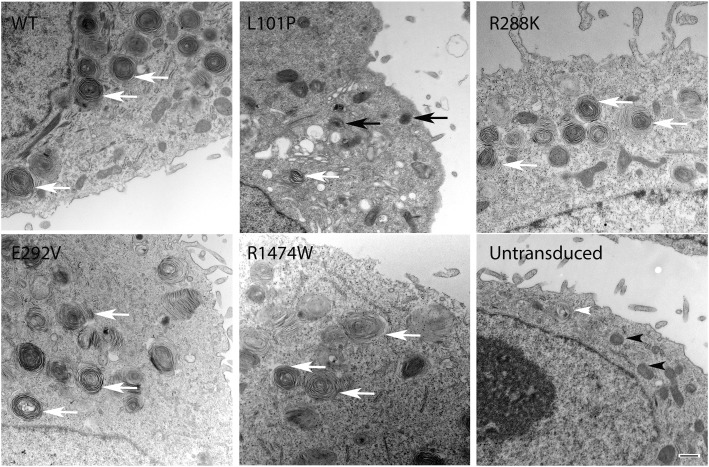
Mutations in the ATP-binding cassette transporter A3 gene (ABCA3) result in severe neonatal respiratory distress syndrome and childhood interstitial lung disease. As most ABCA3 mutations are rare or private, determination of mutation pathogenicity is often based on results from in silico prediction tools, identification in unrelated diseased individuals, statistical association studies, or expert opinion. Functional biologic studies of ABCA3 mutations are needed to confirm mutation pathogenicity and inform clinical decision making. Our objective was to functionally characterize two ABCA3 mutations (p.R288K and p.R1474W) identified among term and late-preterm infants with respiratory distress syndrome with unclear pathogenicity in a genetically versatile model system. We performed transient transfection of HEK293T cells with wild-type or mutant ABCA3 alleles to assess protein processing with immunoblotting. We used transduction of A549 cells with adenoviral vectors, which concurrently silenced endogenous ABCA3 and expressed either wild-type or mutant ABCA3 alleles (p.R288K and p.R1474W) to assess immunofluorescent localization, ATPase activity, and organelle ultrastructure. Both ABCA3 mutations (p.R288K and p.R1474W) encoded proteins with reduced ATPase activity but with normal intracellular localization and protein processing. Ultrastructural phenotypes of lamellar body-like vesicles in A549 cells transduced with mutant alleles were similar to wild type. Mutant proteins encoded by ABCA3 mutations p.R288K and p.R1474W had reduced ATPase activity, a biologically plausible explanation for disruption of surfactant metabolism by impaired phospholipid transport into the lamellar body. These results also demonstrate the usefulness of a genetically versatile, human model system for functional characterization of ABCA3 mutations with unclear pathogenicity.
Keywords: childhood interstitial lung disease; neonatal respiratory distress; respiratory distress syndrome; surfactant.
Figures
References
-
- Fitzgerald ML, Xavier R, Haley KJ, Welti R, Goss JL, Brown CE, Zhuang DZ, Bell SA, Lu N, McKee M, et al. ABCA3 inactivation in mice causes respiratory failure, loss of pulmonary surfactant, and depletion of lung phosphatidylglycerol. J Lipid Res. 2007;48:621–632. - PubMed
-
- Mulugeta S, Gray JM, Notarfrancesco KL, Gonzales LW, Koval M, Feinstein SI, Ballard PL, Fisher AB, Shuman H. Identification of LBM180, a lamellar body limiting membrane protein of alveolar type II cells, as the ABC transporter protein ABCA3. J Biol Chem. 2002;277:22147–22155. - PubMed
-
- Ban N, Matsumura Y, Sakai H, Takanezawa Y, Sasaki M, Arai H, Inagaki N. ABCA3 as a lipid transporter in pulmonary surfactant biogenesis. J Biol Chem. 2007;282:9628–9634. - PubMed
-
- Cheong N, Zhang H, Madesh M, Zhao M, Yu K, Dodia C, Fisher AB, Savani RC, Shuman H. ABCA3 is critical for lamellar body biogenesis in vivo. J Biol Chem. 2007;282:23811–23817. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
