

Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing
- PMID: 27376238
- PMCID: PMC4966644
- DOI: 10.1038/ng.3595
Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing
Abstract
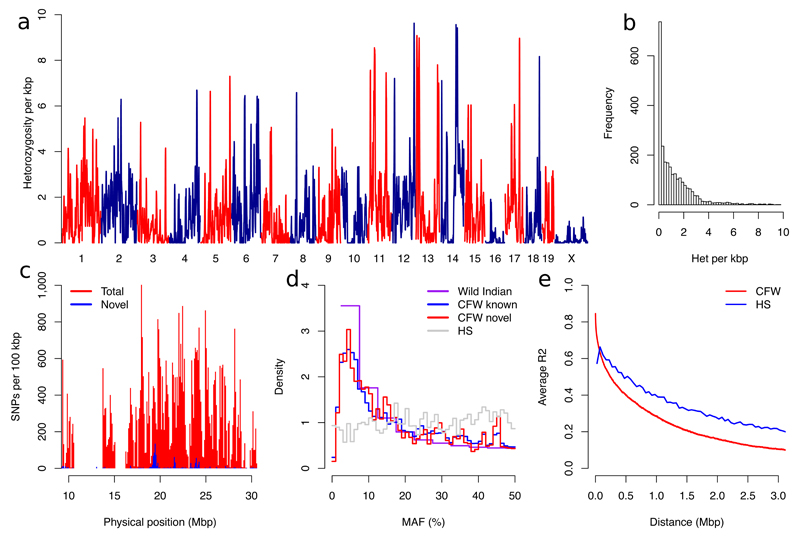
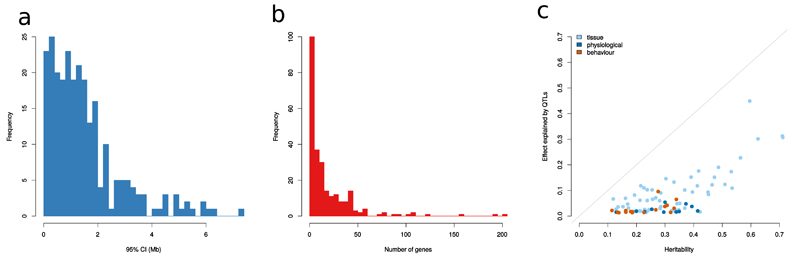
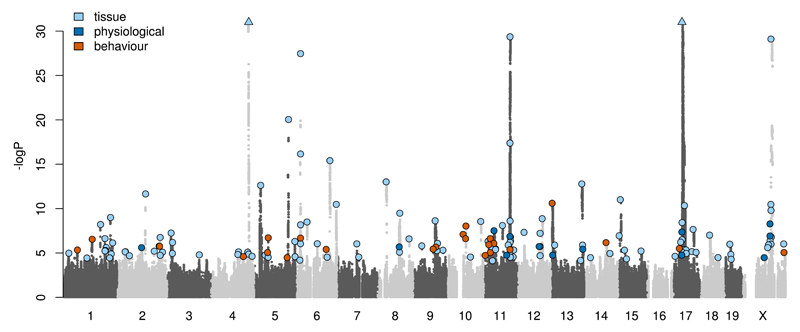
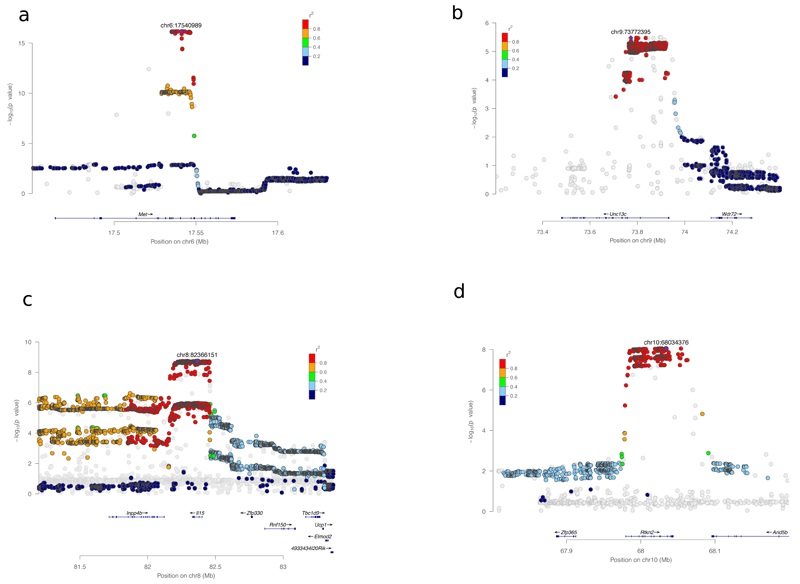
Two bottlenecks impeding the genetic analysis of complex traits in rodents are access to mapping populations able to deliver gene-level mapping resolution and the need for population-specific genotyping arrays and haplotype reference panels. Here we combine low-coverage (0.15×) sequencing with a new method to impute the ancestral haplotype space in 1,887 commercially available outbred mice. We mapped 156 unique quantitative trait loci for 92 phenotypes at a 5% false discovery rate. Gene-level mapping resolution was achieved at about one-fifth of the loci, implicating Unc13c and Pgc1a at loci for the quality of sleep, Adarb2 for home cage activity, Rtkn2 for intensity of reaction to startle, Bmp2 for wound healing, Il15 and Id2 for several T cell measures and Prkca for bone mineral content. These findings have implications for diverse areas of mammalian biology and demonstrate how genome-wide association studies can be extended via low-coverage sequencing to species with highly recombinant outbred populations.
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Huang X, et al. Genome-wide association studies of 14 agronomic traits in rice landraces. Nat Genet. 2010;42:961–7. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
