

Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype
- PMID: 27379212
- PMCID: PMC4909736
- DOI: 10.3389/fonc.2016.00151
Unraveling the Pathogenesis of MDS: The NLRP3 Inflammasome and Pyroptosis Drive the MDS Phenotype
Abstract
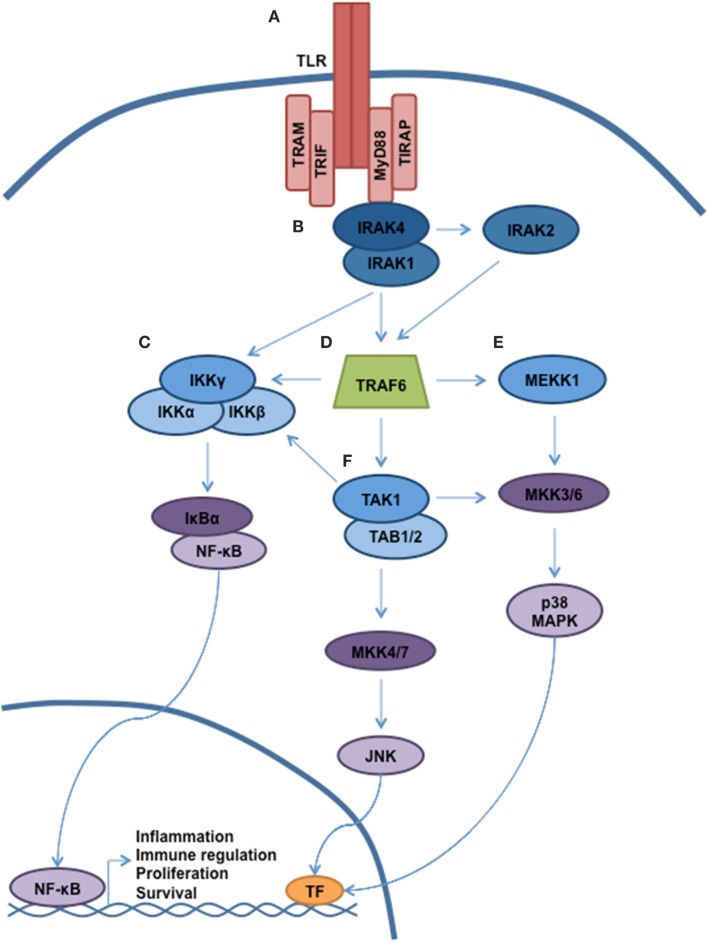
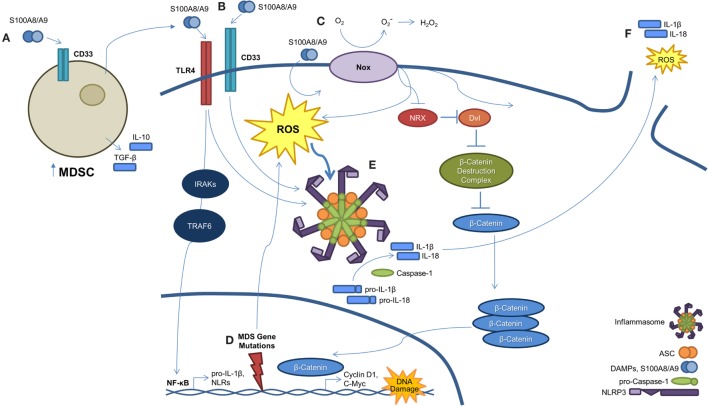
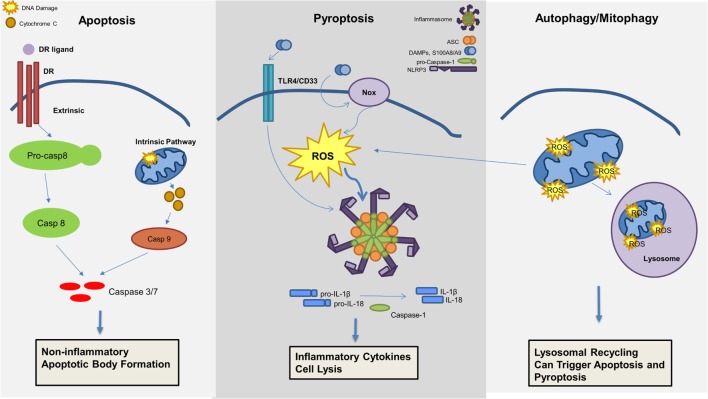
Myelodysplastic syndromes (MDS) are characterized by bone marrow cytological dysplasia and ineffective hematopoiesis in the setting of recurrent somatic gene mutations and chromosomal abnormalities. The underlying pathogenic mechanisms that drive a common clinical phenotype from a diverse array of genetic abnormalities have only recently begun to emerge. Accumulating evidence has highlighted the integral role of the innate immune system in upregulating inflammatory cytokines via NF-κB activation in the pathogenesis of MDS. Recent investigations implicate activation of the NLRP3 inflammasome in hematopoietic stem/progenitor cells as a critical convergence signal in MDS with consequent clonal expansion and pyroptotic cell death though caspase-1 maturation. Specifically, the alarmin S100A9 and/or founder gene mutations trigger pyroptosis through the generation of reactive oxygen species leading to assembly and activation of the redox-sensitive NLRP3 inflammasome and β-catenin, assuring propagation of the MDS clone. More importantly, targeted inhibition of varied steps in this pathway restore effective hematopoiesis. Together, delineation of the role of pyroptosis in the clinical phenotype of MDS patients has identified novel therapeutic strategies that offer significant promise in the treatment of MDS.
Keywords: MDS; NLRP3; S100A9; TLR; inflammasome; pyroptosis.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous