

Rationale for Prolonged Glucocorticoid Use in Pediatric ARDS: What the Adults Can Teach Us
- PMID: 27379217
- PMCID: PMC4906037
- DOI: 10.3389/fped.2016.00058
Rationale for Prolonged Glucocorticoid Use in Pediatric ARDS: What the Adults Can Teach Us
Abstract
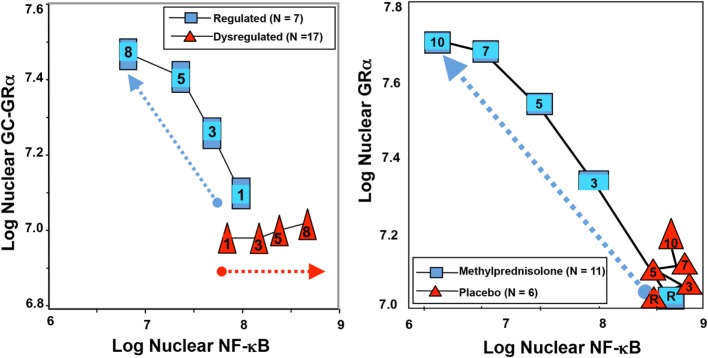
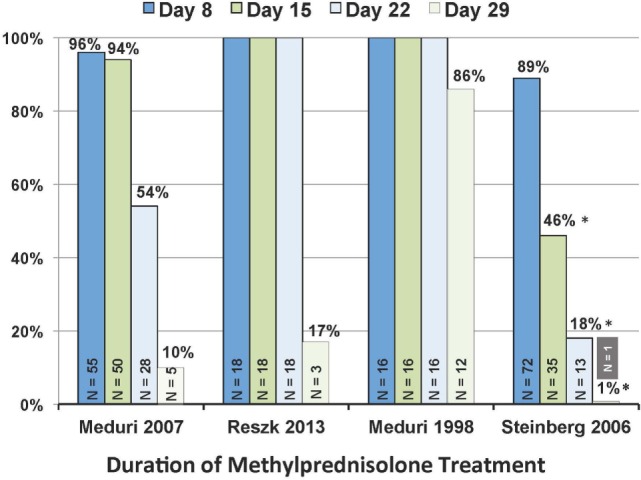
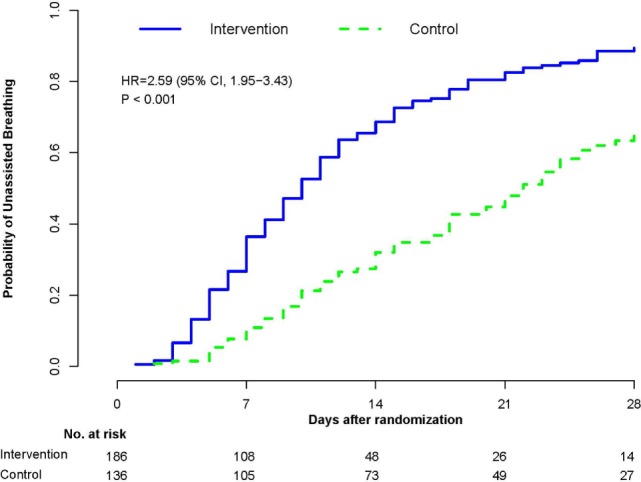
Based on molecular mechanisms and physiologic data, a strong association has been established between dysregulated systemic inflammation and progression of acute respiratory distress syndrome (ARDS). In ARDS patients, glucocorticoid receptor-mediated downregulation of systemic inflammation is essential to restore homeostasis, decrease morbidity and improve survival and can be significantly enhanced with prolonged low-to-moderate dose glucocorticoid treatment. A large body of evidence supports a strong association between prolonged glucocorticoid treatment-induced downregulation of the inflammatory response and improvement in pulmonary and extrapulmonary physiology. The balance of the available data from eight controlled trials (n = 622) provides consistent strong level of evidence for improving patient-centered outcomes and hospital survival. The sizable increase in mechanical ventilation-free days (weighted mean difference, 6.48 days; CI 95% 2.57-10.38, p < 0.0001) and intensive care unit-free days (weighted mean difference, 7.7 days; 95% CI, 3.13-12.20, p < 0.0001) by day 28 is superior to any investigated intervention in ARDS. For treatment initiated before day 14 of ARDS, the increased in hospital survival (70 vs. 52%, OR 2.41, CI 95% 1.50-3.87, p = 0.0003) translates into a number needed to treat to save one life of 5.5. Importantly, prolonged glucocorticoid treatment is not associated with increased risk for nosocomial infections (22 vs. 27%, OR 0.61, CI 95% 0.35-1.04, p = 0.07). Treatment decisions involve a tradeoff between benefits and risks, as well as costs. This low-cost, highly effective therapy is familiar to every physician and has a low risk profile when secondary prevention measures are implemented.
Keywords: acute respiratory distress syndrome; glucocorticoid treatment; mechanical ventilation; survival.
Figures
References
-
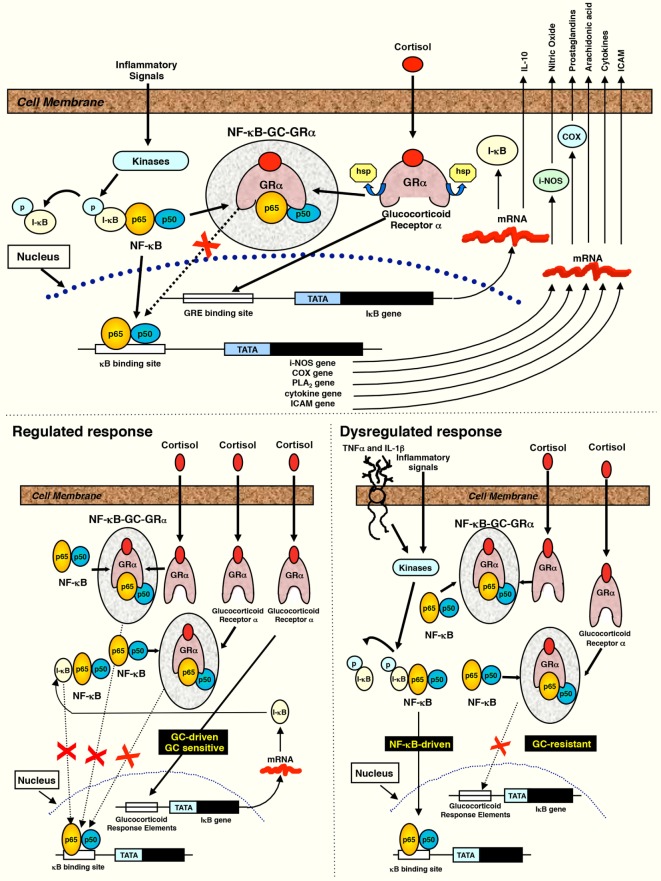
- Meduri GU, Muthiah MP, Carratu P, Eltorky M, Chrousos GP. Nuclear factor-kappaB- and glucocorticoid receptor alpha-mediated mechanisms in the regulation of systemic and pulmonary inflammation during sepsis and acute respiratory distress syndrome. Evidence for inflammation-induced target tissue resistance to glucocorticoids. Neuroimmunomodulation (2005) 12(6):321–38.10.1159/000091126 - DOI - PubMed
Publication types
LinkOut - more resources
Full Text Sources
Other Literature Sources