

Resolving early mesoderm diversification through single-cell expression profiling
- PMID: 27383781
- PMCID: PMC4947525
- DOI: 10.1038/nature18633
Resolving early mesoderm diversification through single-cell expression profiling
Abstract
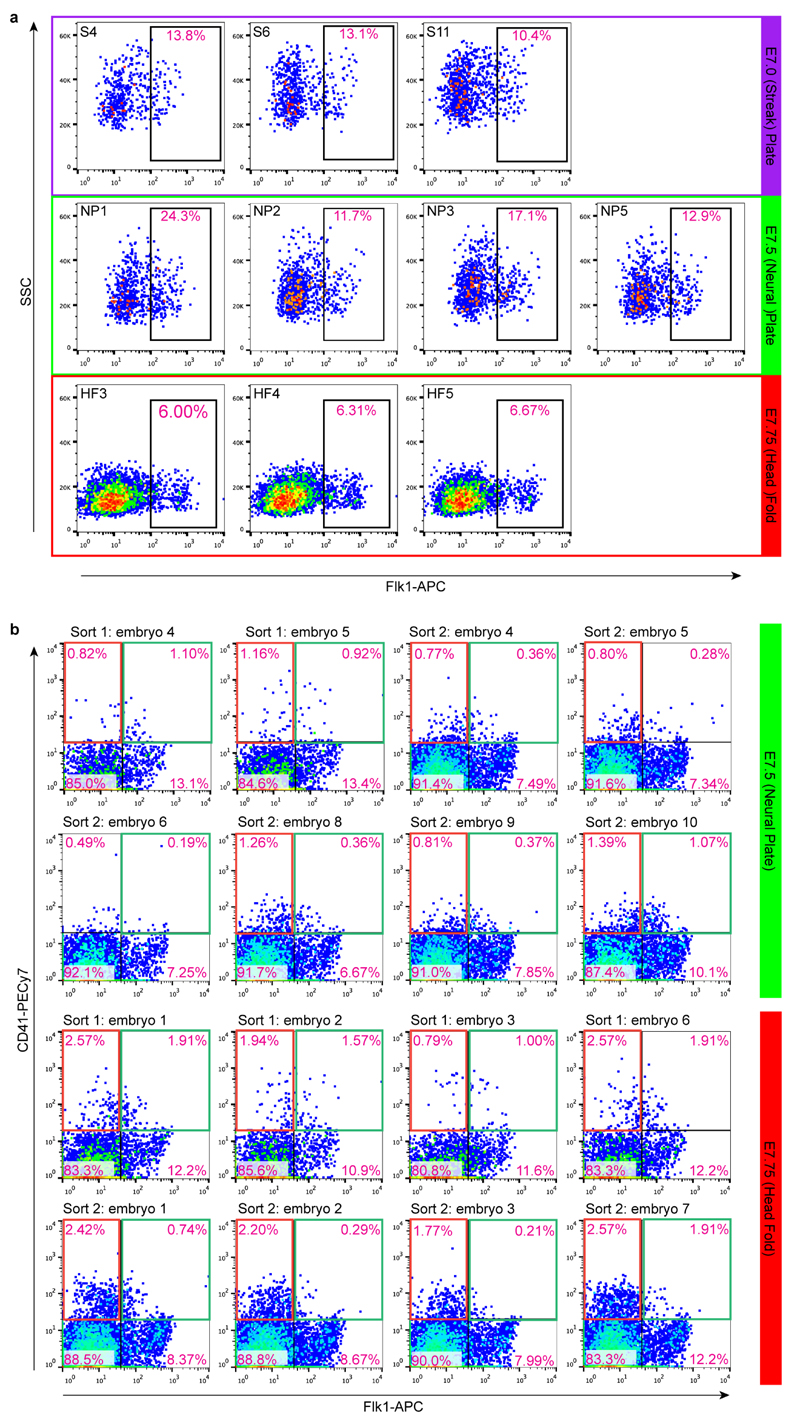
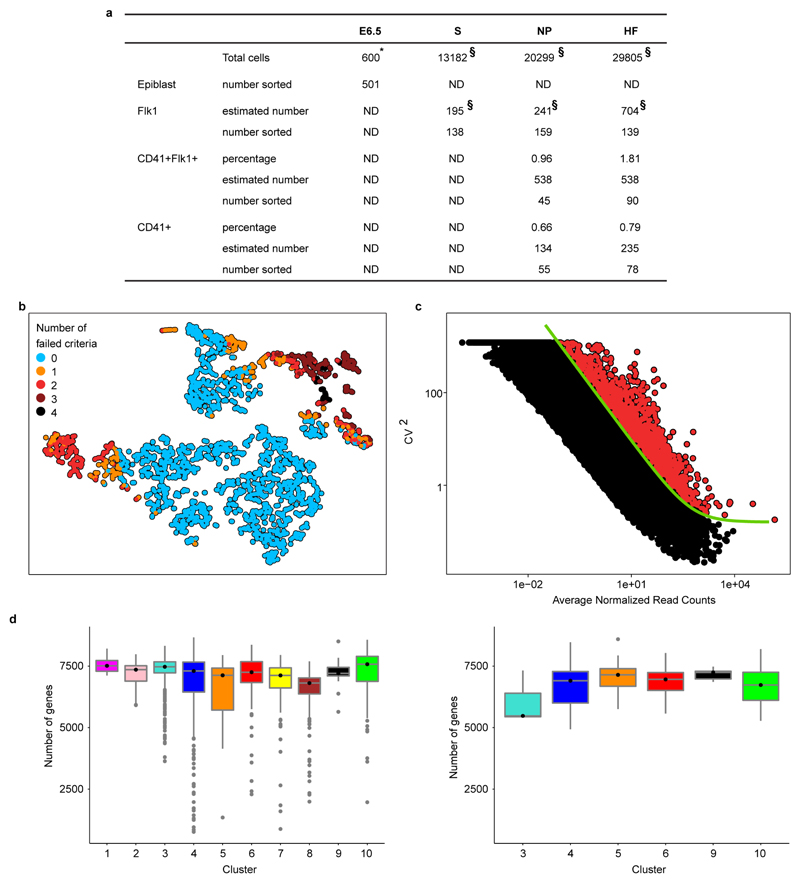
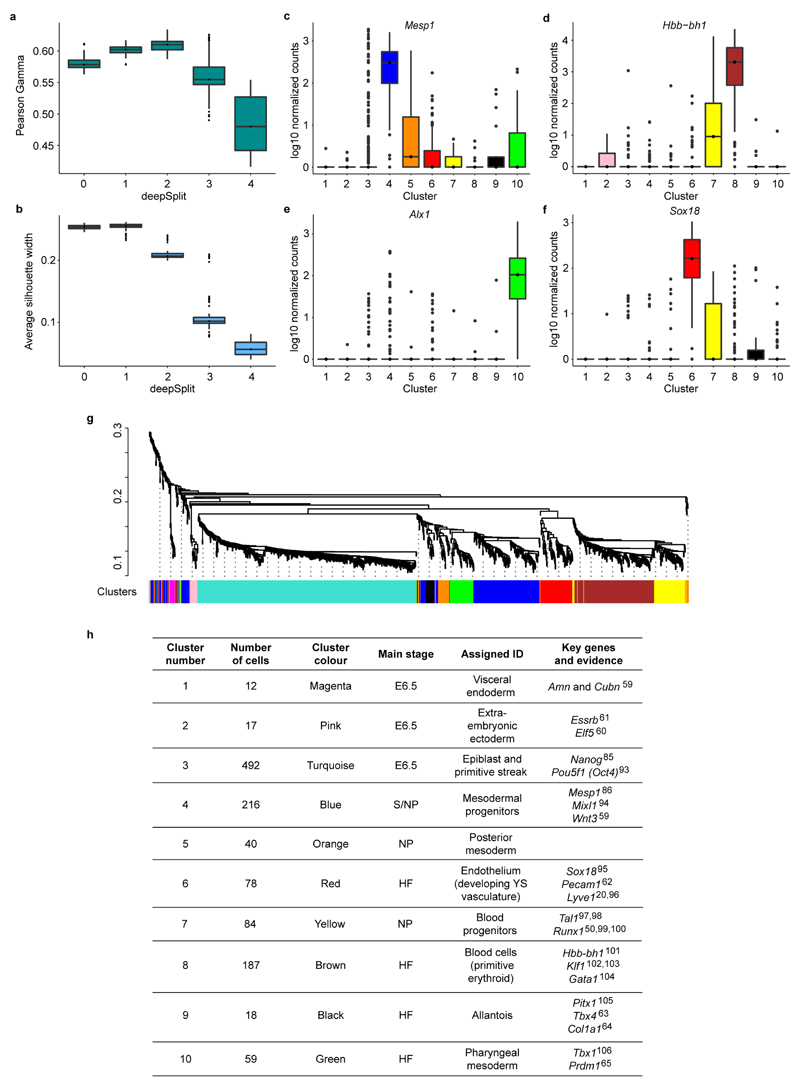
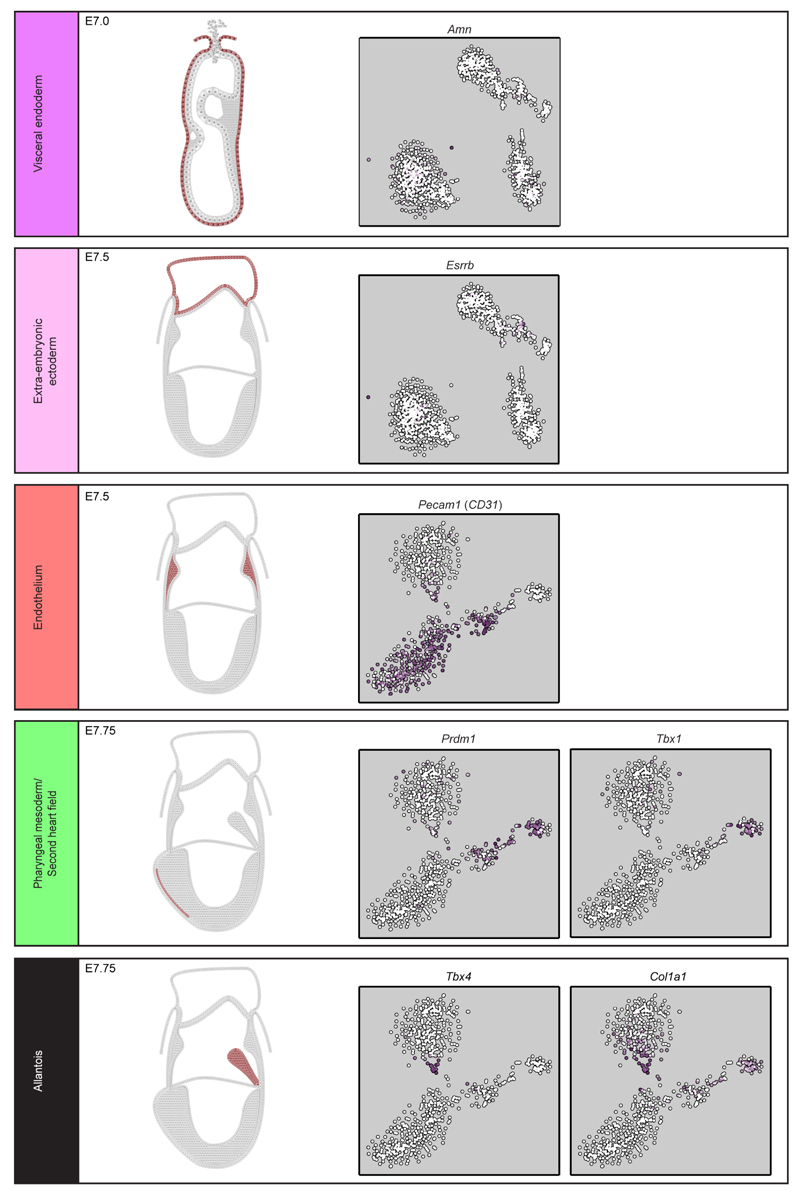
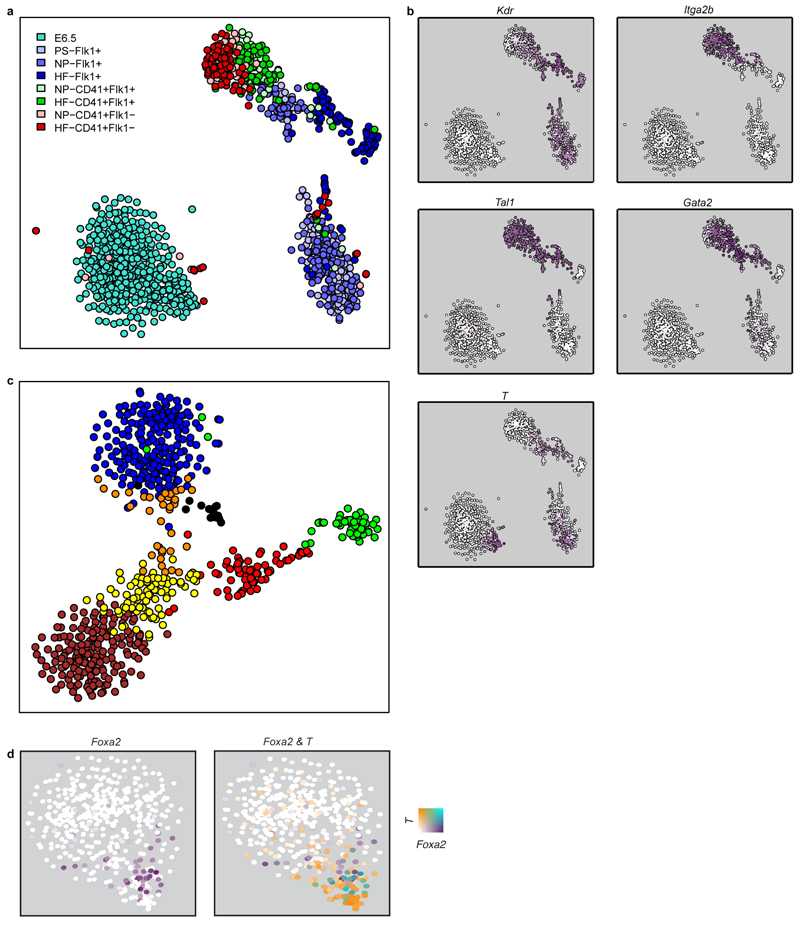
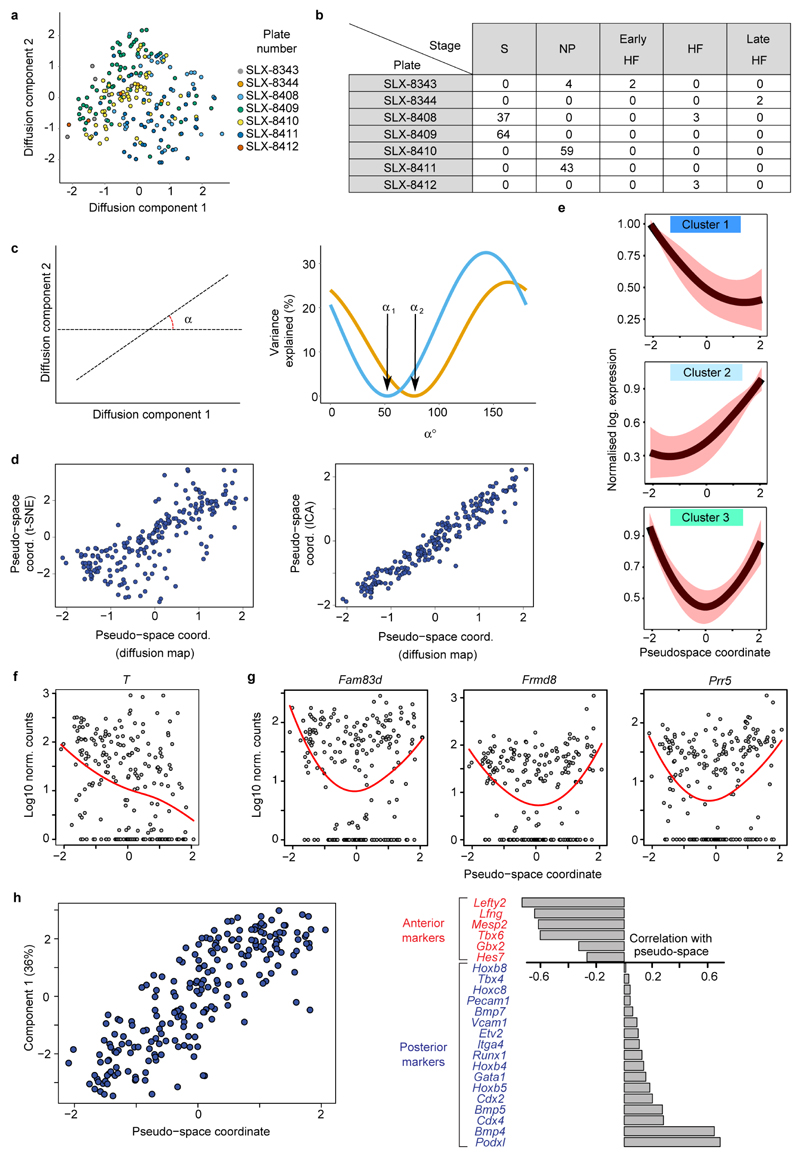
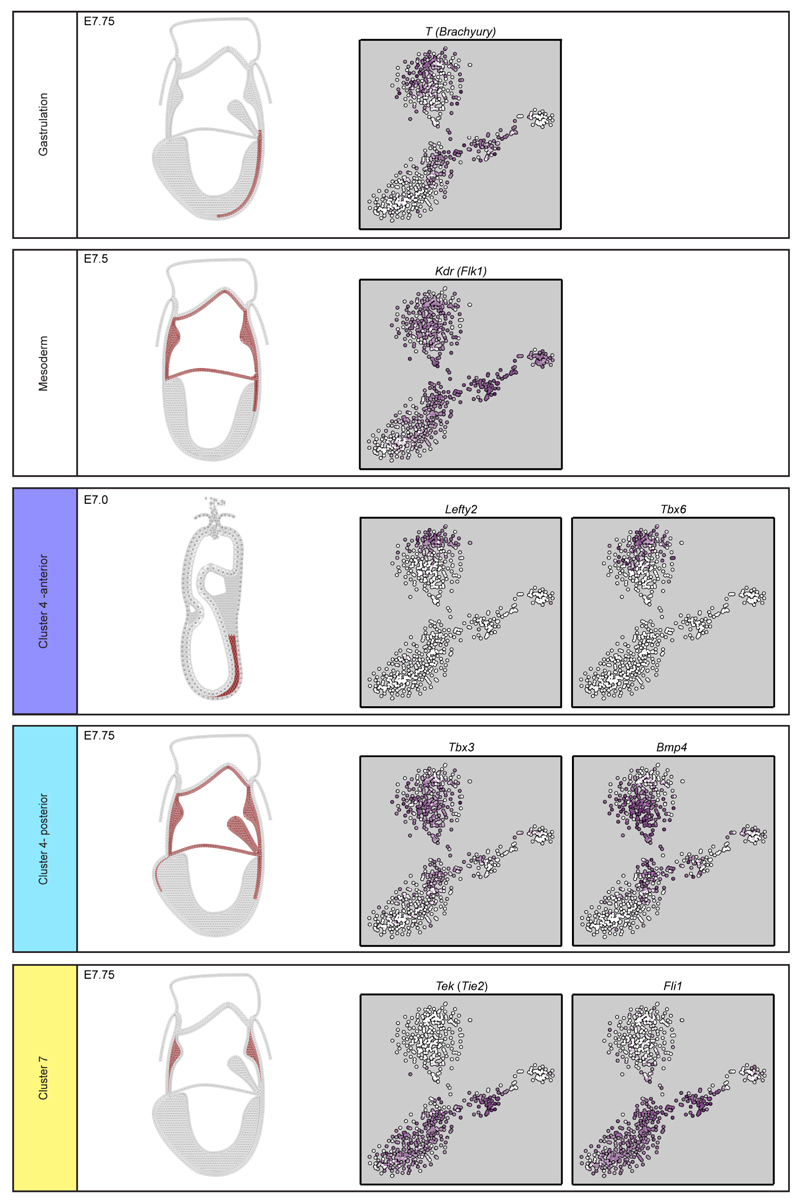
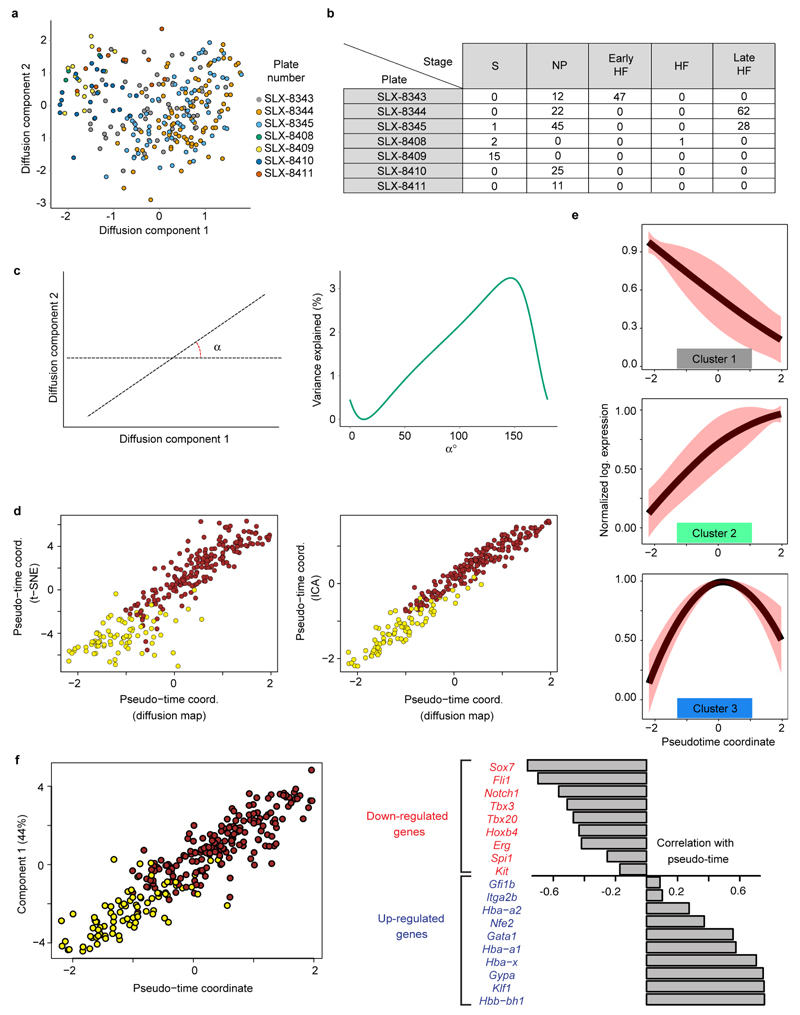
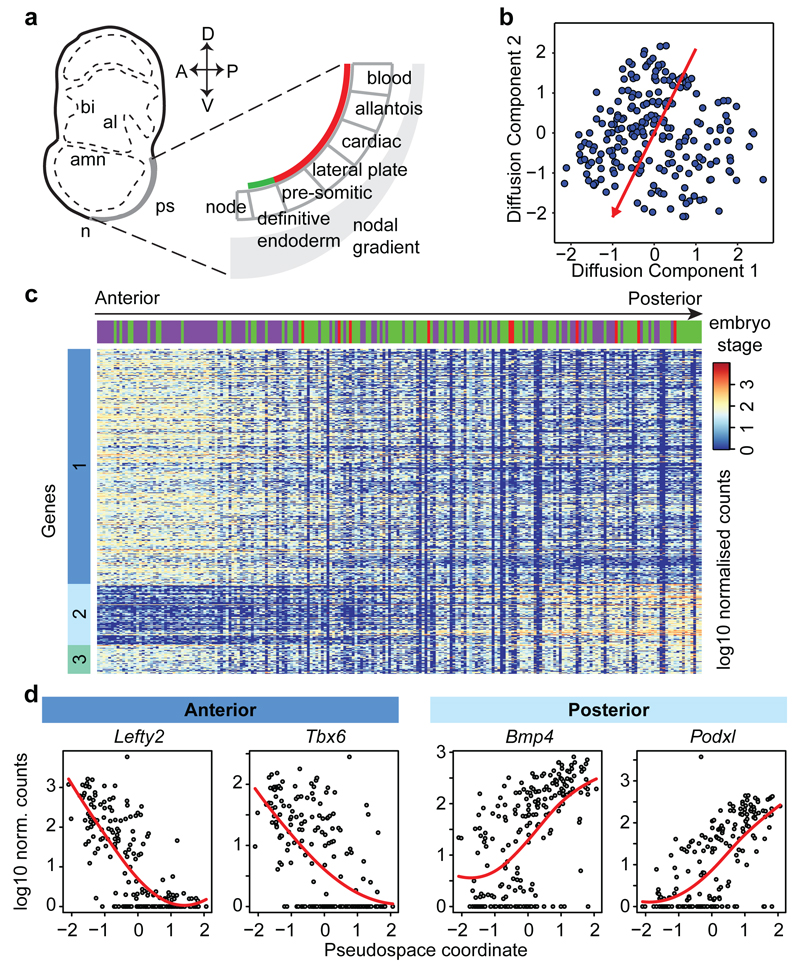
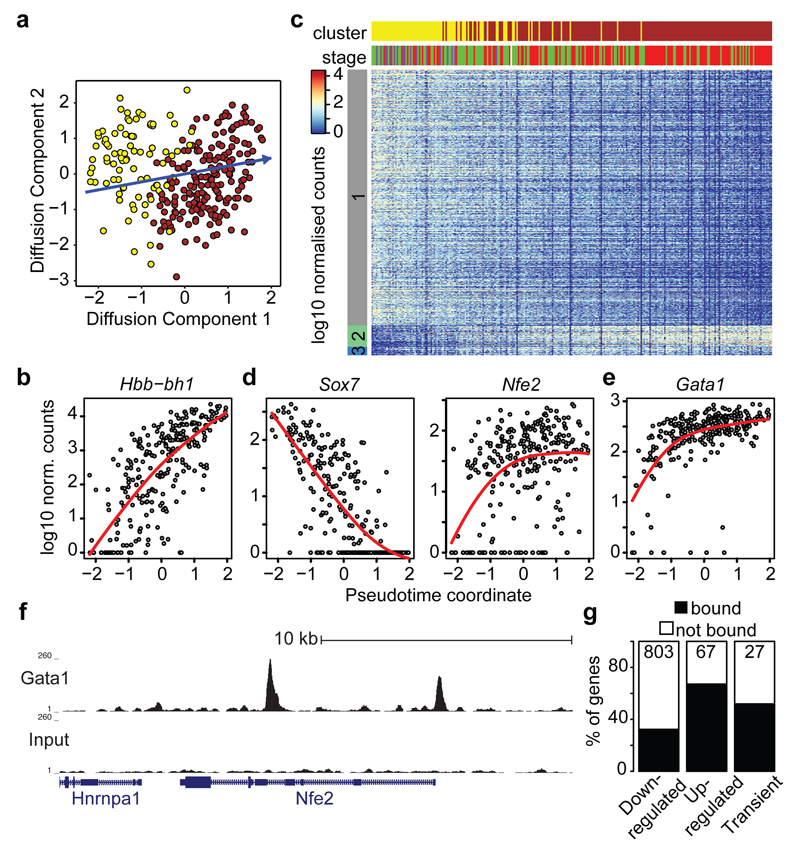
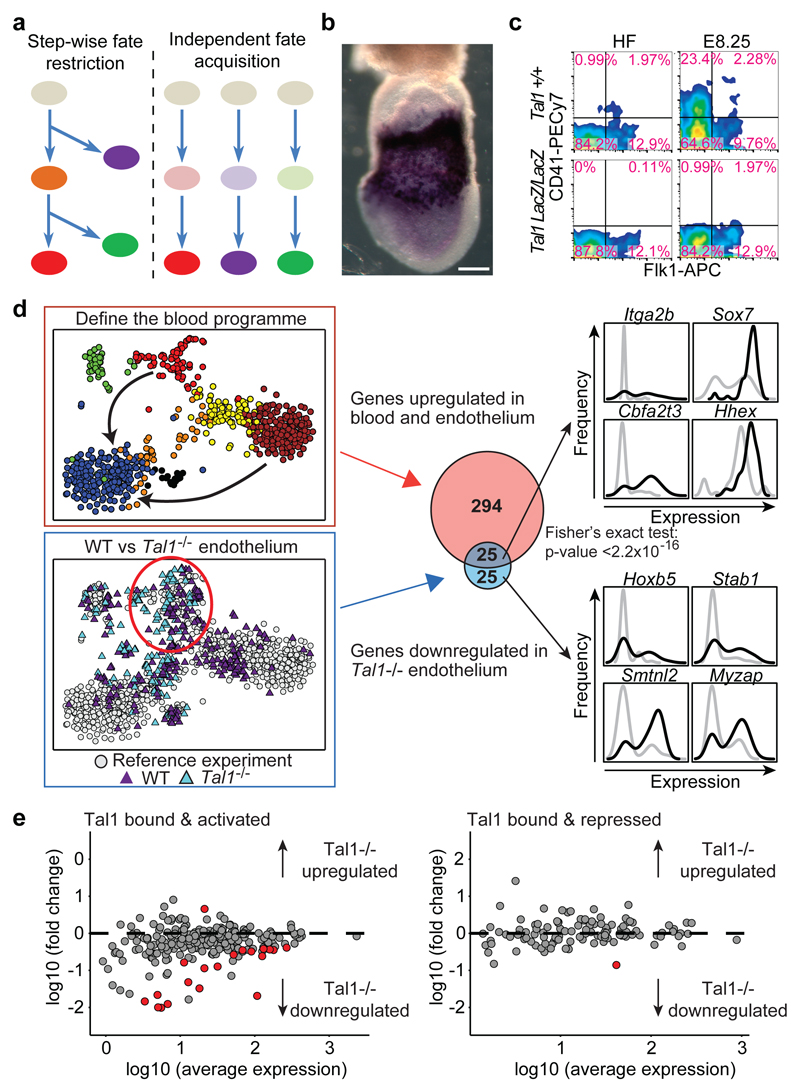
In mammals, specification of the three major germ layers occurs during gastrulation, when cells ingressing through the primitive streak differentiate into the precursor cells of major organ systems. However, the molecular mechanisms underlying this process remain unclear, as numbers of gastrulating cells are very limited. In the mouse embryo at embryonic day 6.5, cells located at the junction between the extra-embryonic region and the epiblast on the posterior side of the embryo undergo an epithelial-to-mesenchymal transition and ingress through the primitive streak. Subsequently, cells migrate, either surrounding the prospective ectoderm contributing to the embryo proper, or into the extra-embryonic region to form the yolk sac, umbilical cord and placenta. Fate mapping has shown that mature tissues such as blood and heart originate from specific regions of the pre-gastrula epiblast, but the plasticity of cells within the embryo and the function of key cell-type-specific transcription factors remain unclear. Here we analyse 1,205 cells from the epiblast and nascent Flk1(+) mesoderm of gastrulating mouse embryos using single-cell RNA sequencing, representing the first transcriptome-wide in vivo view of early mesoderm formation during mammalian gastrulation. Additionally, using knockout mice, we study the function of Tal1, a key haematopoietic transcription factor, and demonstrate, contrary to previous studies performed using retrospective assays, that Tal1 knockout does not immediately bias precursor cells towards a cardiac fate.
Conflict of interest statement
statement The authors declare no competing financial interests.
Figures
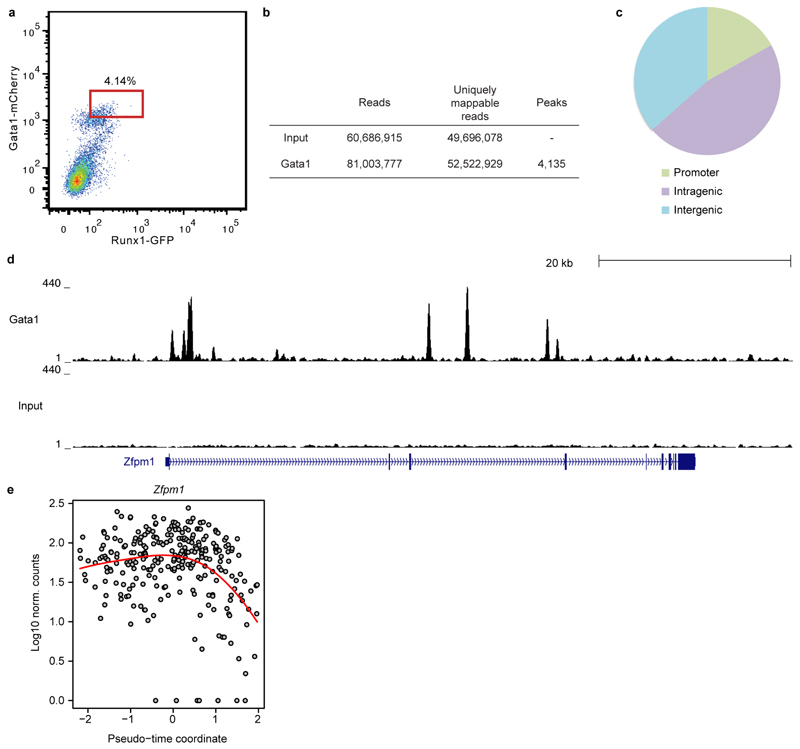
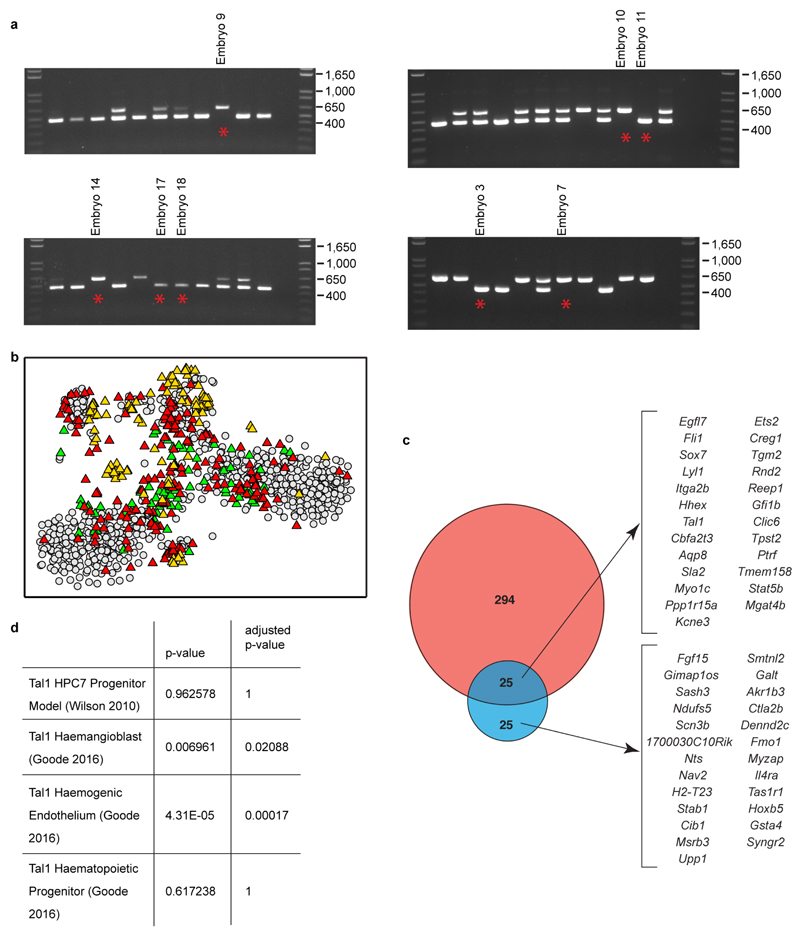
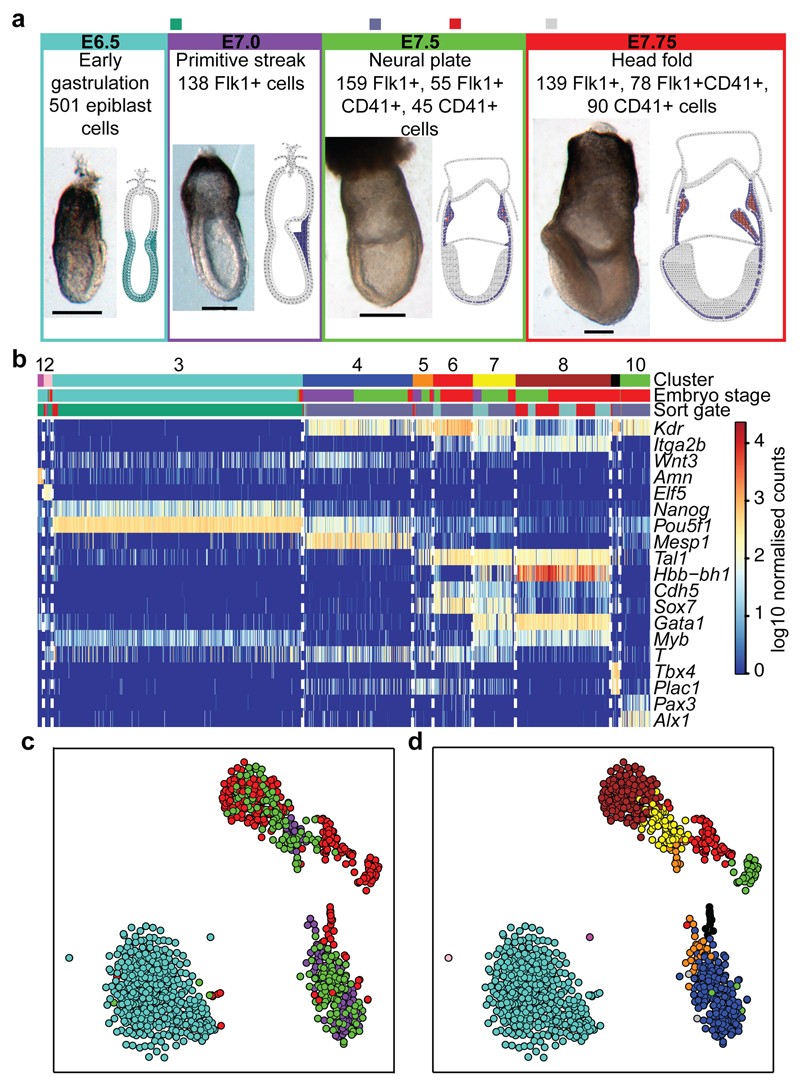
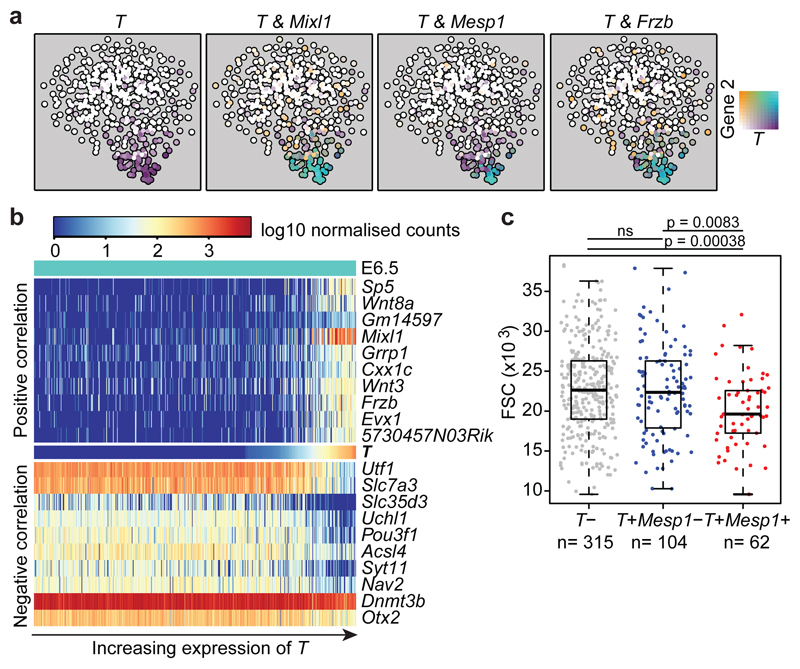
References
-
- Lawson KA, Meneses JJ, Pedersen RA. Clonal analysis of epiblast fate during germ layer formation in the mouse embryo. Development. 1991;113:891–911. - PubMed
-
- Ema M, et al. Primitive erythropoiesis from mesodermal precursors expressing VE-cadherin, PECAM-1, Tie2, endoglin, and CD34 in the mouse embryo. Blood. 2006;108:4018–24. - PubMed
-
- Mikkola HKA, Fujiwara Y, Schlaeger TM, Traver D, Orkin SH. Expression of CD41 marks the initiation of definitive hematopoiesis in the mouse embryo. Blood. 2003;101:508–16. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
