

Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders
- PMID: 27384562
- PMCID: PMC4964441
- DOI: 10.3390/ijms17071065
Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders
Erratum in
-
Coutinho et al. Less Is More: Substrate Reduction Therapy for Lysosomal Storage Disorders. Int. J. Mol. Sci. 2016, 17, 1065.Int J Mol Sci. 2017 Jan 17;18(1):178. doi: 10.3390/ijms18010178. Int J Mol Sci. 2017. PMID: 28106730 Free PMC article.
Abstract
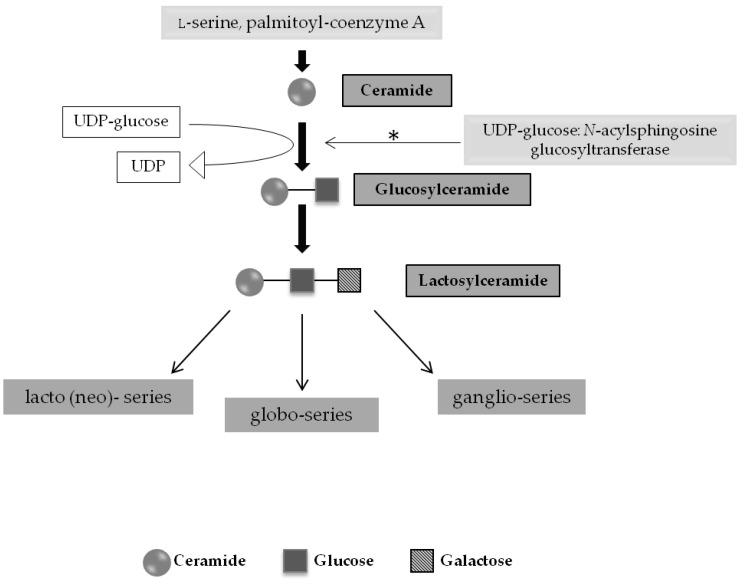
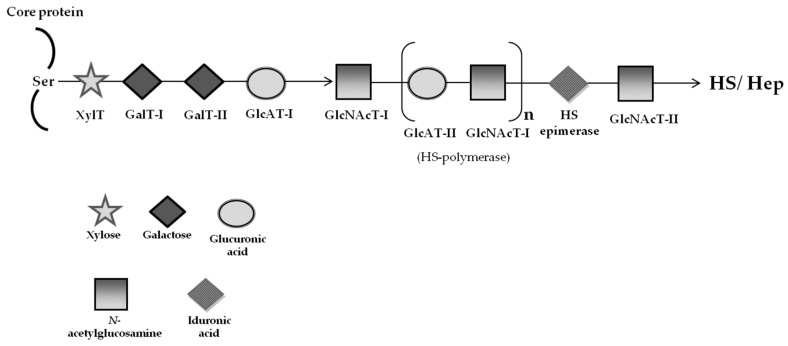
Lysosomal storage diseases (LSDs) are a group of rare, life-threatening genetic disorders, usually caused by a dysfunction in one of the many enzymes responsible for intralysosomal digestion. Even though no cure is available for any LSD, a few treatment strategies do exist. Traditionally, efforts have been mainly targeting the functional loss of the enzyme, by injection of a recombinant formulation, in a process called enzyme replacement therapy (ERT), with no impact on neuropathology. This ineffectiveness, together with its high cost and lifelong dependence is amongst the main reasons why additional therapeutic approaches are being (and have to be) investigated: chaperone therapy; gene enhancement; gene therapy; and, alternatively, substrate reduction therapy (SRT), whose aim is to prevent storage not by correcting the original enzymatic defect but, instead, by decreasing the levels of biosynthesis of the accumulating substrate(s). Here we review the concept of substrate reduction, highlighting the major breakthroughs in the field and discussing the future of SRT, not only as a monotherapy but also, especially, as complementary approach for LSDs.
Keywords: Gaucher disease (GD); Niemann-Pick type C (NPC); Sanfilippo syndrome); combination therapy; eligluistat tartrate; genistein; miglustat; mucopolysaccharidosis type III (MPS III; substrate reduction therapy (SRT).
Figures
Similar articles
-
Substrate reduction therapy.Acta Paediatr. 2008 Apr;97(457):88-93. doi: 10.1111/j.1651-2227.2008.00656.x. Acta Paediatr. 2008. PMID: 18339196 Review.
-
[Current therapeutic strategies in lysosomal disorders].Presse Med. 2014 Nov;43(11):1174-84. doi: 10.1016/j.lpm.2013.12.022. Epub 2014 May 23. Presse Med. 2014. PMID: 24863660 Review. French.
-
Substrate deprivation therapy: a new hope for patients suffering from neuronopathic forms of inherited lysosomal storage diseases.J Appl Genet. 2007;48(4):383-8. doi: 10.1007/BF03195237. J Appl Genet. 2007. PMID: 17998597 Review.
-
Substrate reduction therapy of glycosphingolipid storage disorders.J Inherit Metab Dis. 2006 Apr-Jun;29(2-3):449-56. doi: 10.1007/s10545-006-0272-5. J Inherit Metab Dis. 2006. PMID: 16763917
-
Pulmonary involvement in selected lysosomal storage diseases and the impact of enzyme replacement therapy: A state-of-the art review.Clin Respir J. 2020 May;14(5):422-429. doi: 10.1111/crj.13150. Epub 2020 Jan 22. Clin Respir J. 2020. PMID: 31912638 Review.
Cited by
-
Venglustat, an orally administered glucosylceramide synthase inhibitor: Assessment over 3 years in adult males with classic Fabry disease in an open-label phase 2 study and its extension study.Mol Genet Metab. 2023 Feb;138(2):106963. doi: 10.1016/j.ymgme.2022.11.002. Epub 2022 Nov 9. Mol Genet Metab. 2023. PMID: 36481125 Free PMC article. Clinical Trial.
-
Biochemical characteristics of point mutated Capra hircus lysosome α-mannosidase.J Vet Med Sci. 2023 Feb 21;85(2):244-251. doi: 10.1292/jvms.22-0222. Epub 2023 Jan 2. J Vet Med Sci. 2023. PMID: 36596563 Free PMC article.
-
Extracellular Vesicles as Tools for Crossing the Blood-Brain Barrier to Treat Lysosomal Storage Diseases.Life (Basel). 2025 Jan 9;15(1):70. doi: 10.3390/life15010070. Life (Basel). 2025. PMID: 39860010 Free PMC article. Review.
-
Mutation Spectrum of GAA Gene in Pompe Disease: Current Knowledge and Results of an Italian Study.Int J Mol Sci. 2024 Aug 23;25(17):9139. doi: 10.3390/ijms25179139. Int J Mol Sci. 2024. PMID: 39273088 Free PMC article.
-
In vitro substrate reduction, chaperone and immunomodulation treatments reduce heparan sulfate in mucolipidosis III human fibroblasts.Genet Mol Biol. 2023 Dec 4;46(3 Suppl 1):e20230117. doi: 10.1590/1678-4685-GMB-2023-0117. eCollection 2023. Genet Mol Biol. 2023. PMID: 38047750 Free PMC article.
References
-
- Hers H.G. Inborn lysosomal diseases. Gastroenterology. 1965;48:625–633. - PubMed
-
- De Duve C. From lysosomes to storage diseases and back: A personal reminiscence. In: Barranger J.A., Cabrera-Salazar M., editors. Lysosomal Storage Disorders. Springer US; New York, NY, USA: 2007. pp. 1–5.
-
- Deduve C. From cytases to lysosomes. Fed. Proc. 1964;23:1045–1049. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
