

Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes
- PMID: 27385709
- PMCID: PMC5035407
- DOI: 10.1128/JCM.00330-16
Comparison of Next-Generation Sequencing Technologies for Comprehensive Assessment of Full-Length Hepatitis C Viral Genomes
Abstract
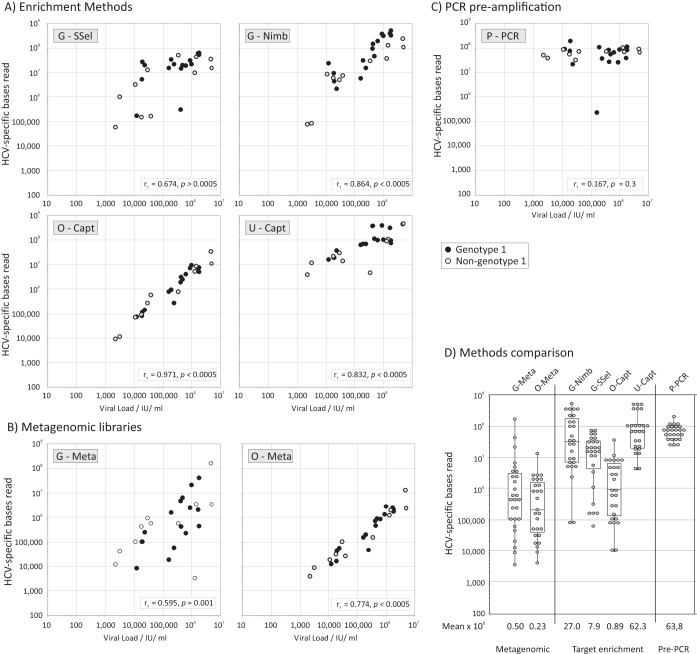
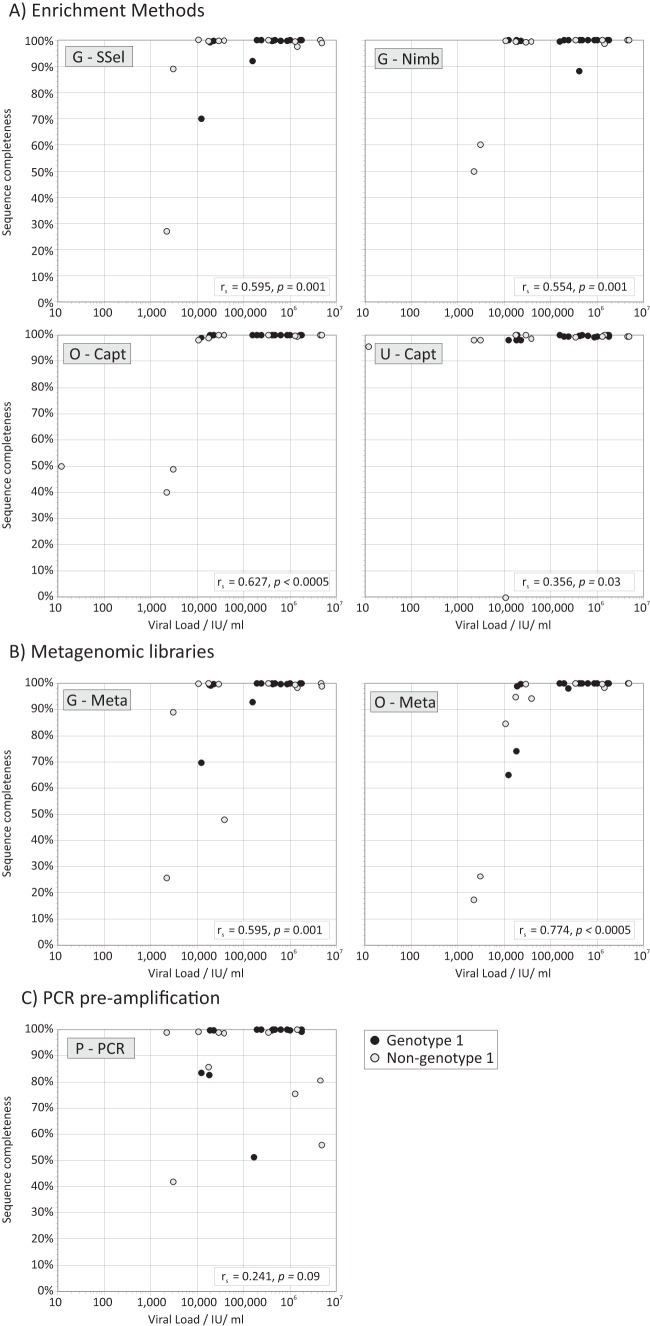
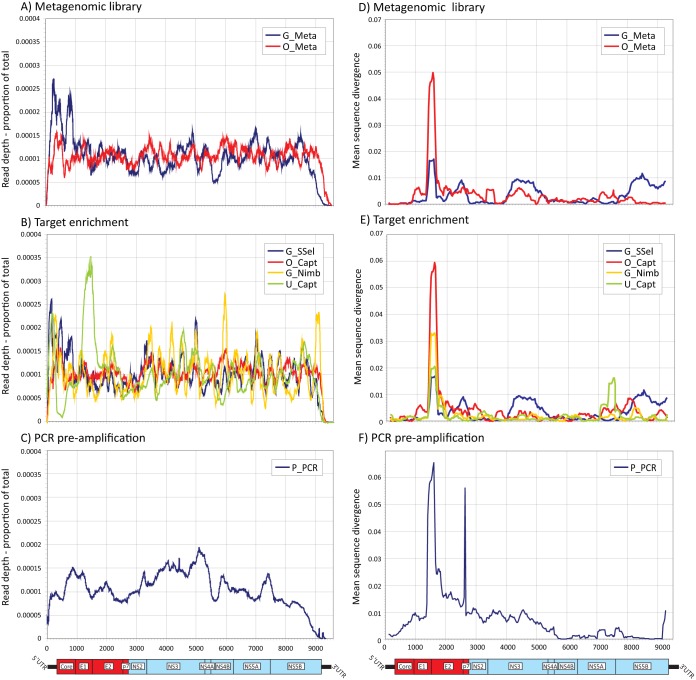
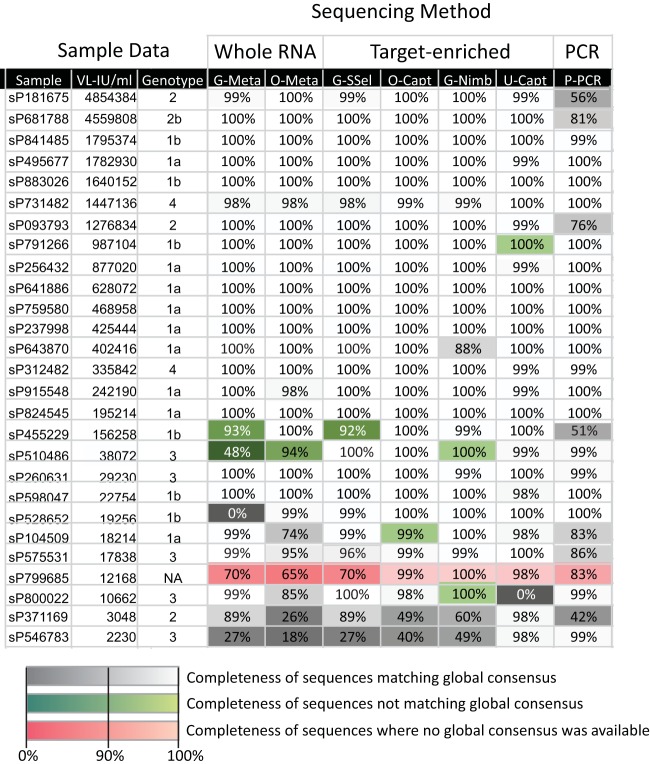
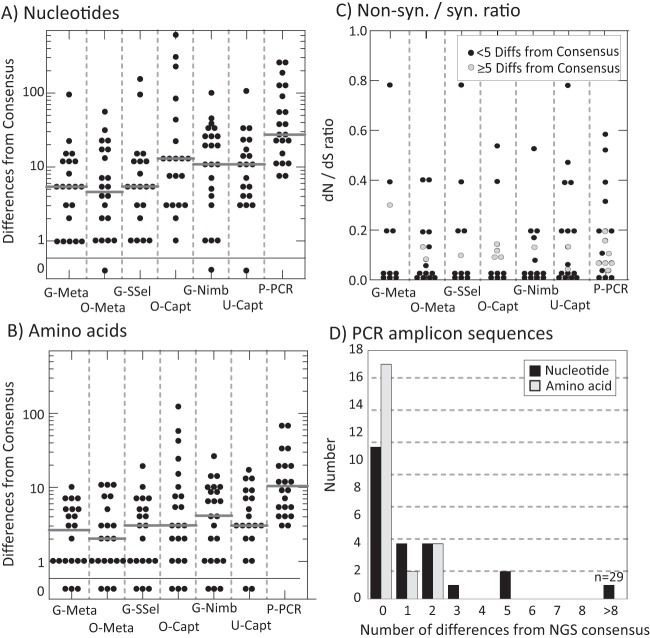
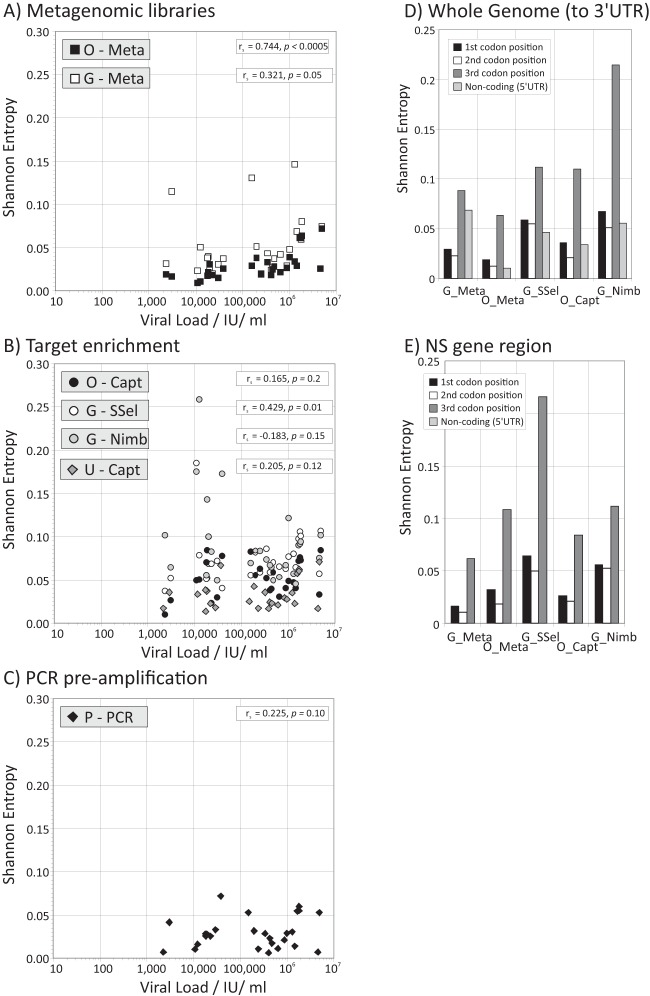
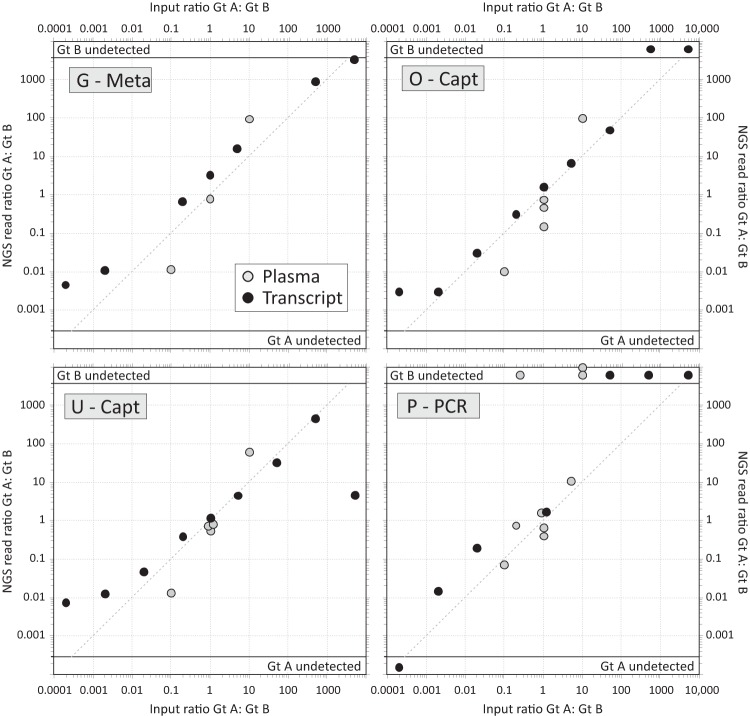
Affordable next-generation sequencing (NGS) technologies for hepatitis C virus (HCV) may potentially identify both viral genotype and resistance genetic motifs in the era of directly acting antiviral (DAA) therapies. This study compared the ability of high-throughput NGS methods to generate full-length, deep, HCV sequence data sets and evaluated their utility for diagnostics and clinical assessment. NGS methods using (i) unselected HCV RNA (metagenomics), (ii) preenrichment of HCV RNA by probe capture, and (iii) HCV preamplification by PCR implemented in four United Kingdom centers were compared. Metrics of sequence coverage and depth, quasispecies diversity, and detection of DAA resistance-associated variants (RAVs), mixed HCV genotypes, and other coinfections were compared using a panel of samples with different viral loads, genotypes, and mixed HCV genotypes/subtypes [geno(sub)types]. Each NGS method generated near-complete genome sequences from more than 90% of samples. Enrichment methods and PCR preamplification generated greater sequence depth and were more effective for samples with low viral loads. All NGS methodologies accurately identified mixed HCV genotype infections. Consensus sequences generated by different NGS methods were generally concordant, and majority RAVs were consistently detected. However, methods differed in their ability to detect minor populations of RAVs. Metagenomic methods identified human pegivirus coinfections. NGS provided a rapid, inexpensive method for generating whole HCV genomes to define infecting genotypes, RAVs, comprehensive viral strain analysis, and quasispecies diversity. Enrichment methods are particularly suited for high-throughput analysis while providing the genotype and information on potential DAA resistance.
Copyright © 2016 Thomson et al.
Figures


Comment in
-
Next-Generation Sequencing: a Diagnostic One-Stop Shop for Hepatitis C?J Clin Microbiol. 2016 Oct;54(10):2427-30. doi: 10.1128/JCM.01423-16. Epub 2016 Aug 10. J Clin Microbiol. 2016. PMID: 27510828 Free PMC article.
References
-
- Jacobson IM, Dore GJ, Foster GR, Fried MW, Radu M, Rafalsky VV, Moroz L, Craxi A, Peeters M, Lenz O, Ouwerkerk-Mahadevan S, De La Rosa G, Kalmeijer R, Scott J, Sinha R, Beumont-Mauviel M. 2014. Simeprevir with pegylated interferon alfa 2a plus ribavirin in treatment-naive patients with chronic hepatitis C virus genotype 1 infection (QUEST-1): a phase 3, randomised, double-blind, placebo-controlled trial. Lancet 384:403–413. doi:10.1016/S0140-6736(14)60494-3. - DOI - PubMed
-
- Kowdley KV, Lawitz E, Crespo I, Hassanein T, Davis MN, DeMicco M, Bernstein DE, Afdhal N, Vierling JM, Gordon SC, Anderson JK, Hyland RH, Dvory-Sobol H, An D, Hindes RG, Albanis E, Symonds WT, Berrey MM, Nelson DR, Jacobson IM. 2013. Sofosbuvir with pegylated interferon alfa-2a and ribavirin for treatment-naive patients with hepatitis C genotype-1 infection (ATOMIC): an open-label, randomised, multicentre phase 2 trial. Lancet 381:2100–2107. doi:10.1016/S0140-6736(13)60247-0. - DOI - PubMed
-
- Lawitz E, Poordad FF, Pang PS, Hyland RH, Ding X, Mo H, Symonds WT, McHutchison JG, Membreno FE. 2014. Sofosbuvir and ledipasvir fixed-dose combination with and without ribavirin in treatment-naive and previously treated patients with genotype 1 hepatitis C virus infection (LONESTAR): an open-label, randomised, phase 2 trial. Lancet 383:515–523. doi:10.1016/S0140-6736(13)62121-2. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
- MC_UU_12014/1/MRC_/Medical Research Council/United Kingdom
- G0801822/MRC_/Medical Research Council/United Kingdom
- 14/02/17/DH_/Department of Health/United Kingdom
- C0365/MRF_/MRF_/United Kingdom
- MR/K010239/1/MRC_/Medical Research Council/United Kingdom
- 091663/WT_/Wellcome Trust/United Kingdom
- MR/K01532X/1/MRC_/Medical Research Council/United Kingdom
- MC_EX_UU_G1000717/MRC_/Medical Research Council/United Kingdom
- 109965/Z/15/Z/WT_/Wellcome Trust/United Kingdom
- U19 AI082630/AI/NIAID NIH HHS/United States
- MC_UU_12014/12/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
