

Characteristics of safety information obtained from postmarketing observational studies for re-examination in Japan
- PMID: 27386350
- PMCID: PMC4923017
- DOI: 10.1186/s40064-016-2365-4
Characteristics of safety information obtained from postmarketing observational studies for re-examination in Japan
Abstract
Background: In Japan, postmarketing surveillance (PMS) studies are required for newly approved drug products to further collect safety information in clinical settings. "PMS study" is a general term encompassing both postmarketing observational (PMO) studies and postmarketing intervention studies for re-examination. Each PMS study is conducted under contracts between the pharmaceutical company and medical institutions in accordance with Good Postmarketing Study Practice. It has been reported that the safety information collected postmarketing is limited because of underreporting. The objective of this investigation was to identify differences among profiles of the drug product safety information collected through intervention studies and observational studies before and after approval. Our study addressed whether the issue of underreporting, generally considered as associated with observational studies, occurs in PMO studies for re-examination. In addition, we considered potential causes of such underreporting.
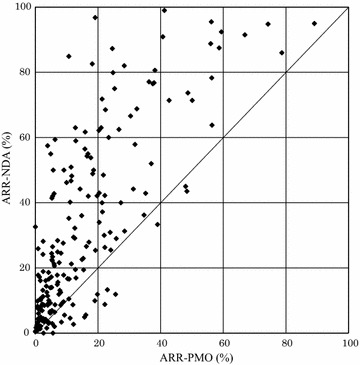
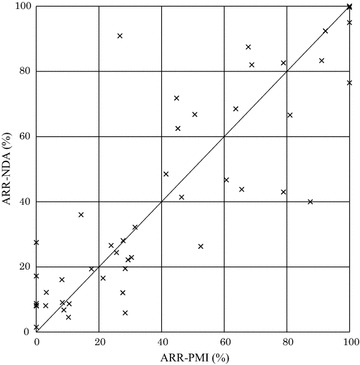
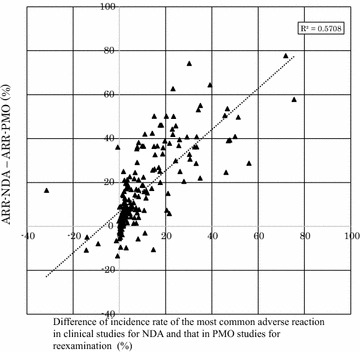
Results: The overall adverse reaction rate was lower in PMO studies than in intervention studies before approval in almost all cases. The adverse reaction rate in intervention studies exhibited similar profiles regardless of whether they were conducted prior to or following approval. In addition, we found that one reason for a lower adverse reaction rate in PMO studies was that the number of reports of adverse reactions that had occurred frequently prior to approval decreased postmarketing.
Conclusions: Underreporting was observed even in PMO studies for re-examination under the Japanese regulation. Although it was suggested that expected and common adverse reactions were more likely to be subject to underreporting, further investigation is warranted to explore the reasons for the under-reporting in PMO studies.
Keywords: Package insert; Pharmacovigilance; Postmarketing surveillance; Re-examination; Safety.
Figures
References
-
- Avery AJ, Anderson C, Bond CM, Fortnum H, Gifford A, Hannaford PC, et al. Evaluation of patient reporting of adverse drug reactions to the UK “Yellow Card Scheme”: literature review, descriptive and qualitative analyses, and questionnaire surveys. Health Technol Assess. 2011;20:1–234. - PubMed
-
- European Union (2011) Volume 9A of the rules governing medicinal products in the European Union—guidelines on pharmacovigilance for medicinal products for human use
LinkOut - more resources
Full Text Sources
Other Literature Sources
