

Identification of Novel and Recurrent Disease-Causing Mutations in Retinal Dystrophies Using Whole Exome Sequencing (WES): Benefits and Limitations
- PMID: 27391102
- PMCID: PMC4938416
- DOI: 10.1371/journal.pone.0158692
Identification of Novel and Recurrent Disease-Causing Mutations in Retinal Dystrophies Using Whole Exome Sequencing (WES): Benefits and Limitations
Abstract
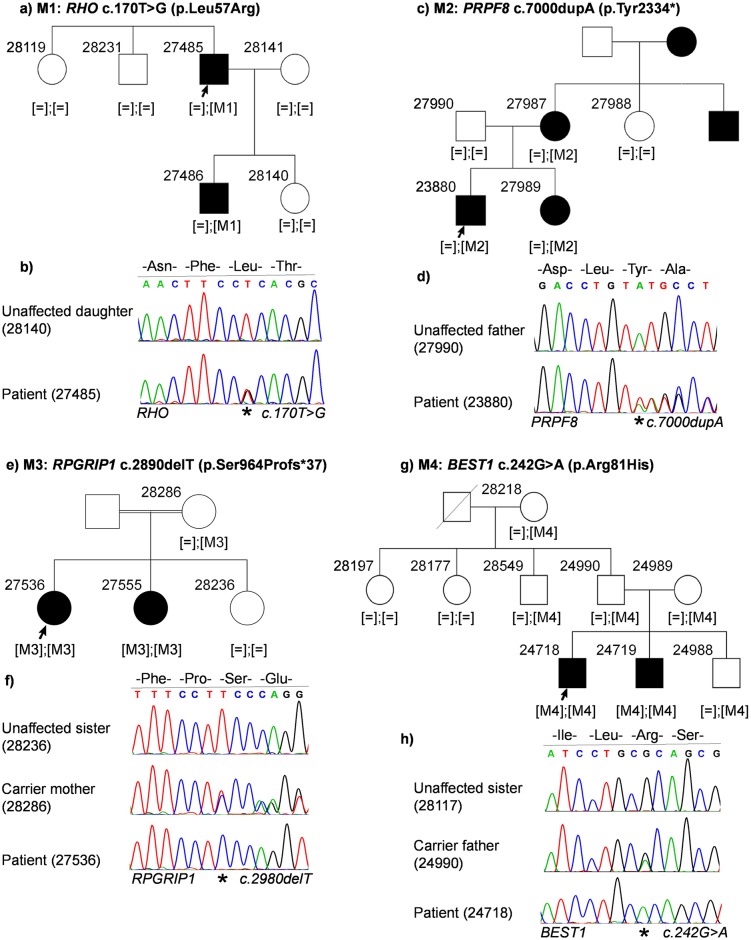
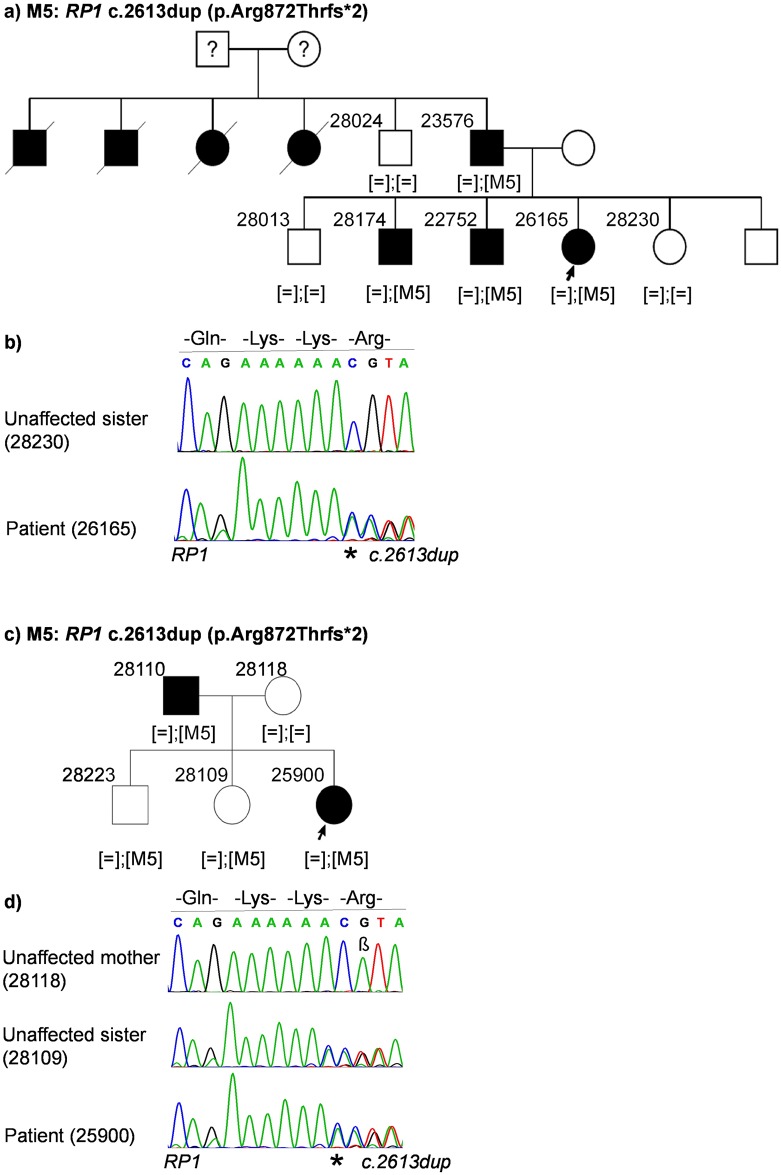
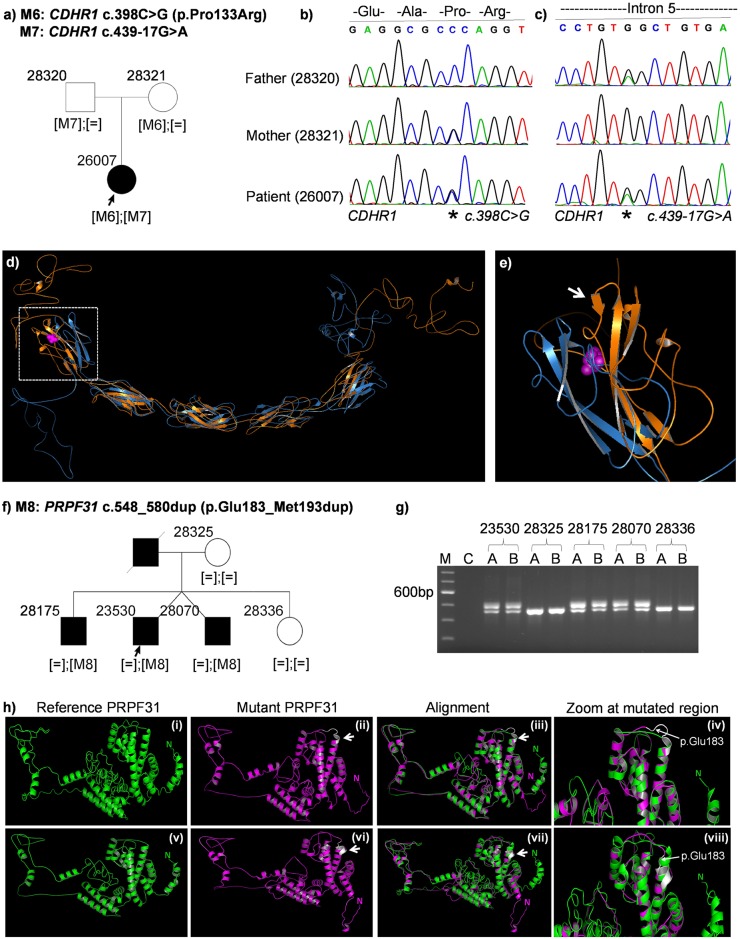
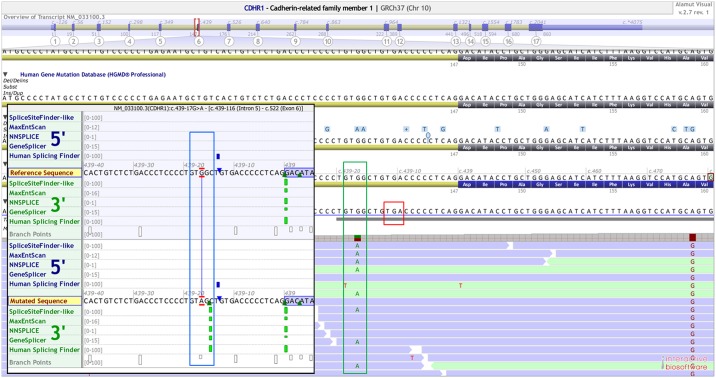
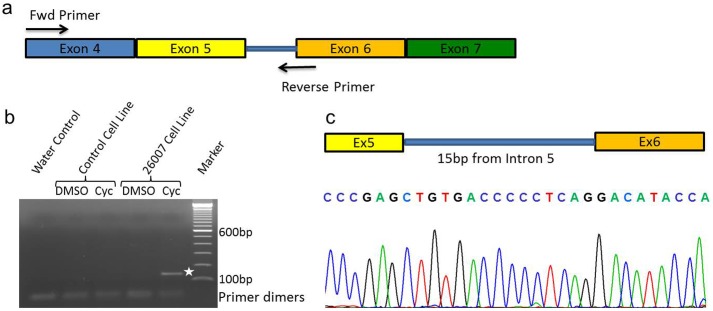
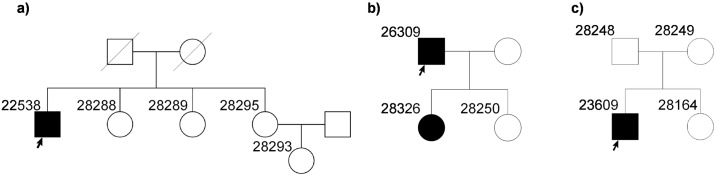
Inherited retinal dystrophies (IRDs) are Mendelian diseases with tremendous genetic and phenotypic heterogeneity. Identification of the underlying genetic basis of these dystrophies is therefore challenging. In this study we employed whole exome sequencing (WES) in 11 families with IRDs and identified disease-causing variants in 8 of them. Sequence analysis of about 250 IRD-associated genes revealed 3 previously reported disease-associated variants in RHO, BEST1 and RP1. We further identified 5 novel pathogenic variants in RPGRIP1 (p.Ser964Profs*37), PRPF8 (p.Tyr2334Leufs*51), CDHR1 (p.Pro133Arg and c.439-17G>A) and PRPF31 (p.Glu183_Met193dup). In addition to confirming the power of WES in genetic diagnosis of IRDs, we document challenges in data analysis and show cases where the underlying genetic causes of IRDs were missed by WES and required additional techniques. For example, the mutation c.439-17G>A in CDHR1 would be rated unlikely applying the standard WES analysis. Only transcript analysis in patient fibroblasts confirmed the pathogenic nature of this variant that affected splicing of CDHR1 by activating a cryptic splice-acceptor site. In another example, a 33-base pair duplication in PRPF31 missed by WES could be identified only via targeted analysis by Sanger sequencing. We discuss the advantages and challenges of using WES to identify mutations in heterogeneous diseases like IRDs.
Conflict of interest statement
Figures
References
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
