

Deep Whole-Genome Sequencing to Detect Mixed Infection of Mycobacterium tuberculosis
- PMID: 27391214
- PMCID: PMC4938208
- DOI: 10.1371/journal.pone.0159029
Deep Whole-Genome Sequencing to Detect Mixed Infection of Mycobacterium tuberculosis
Abstract
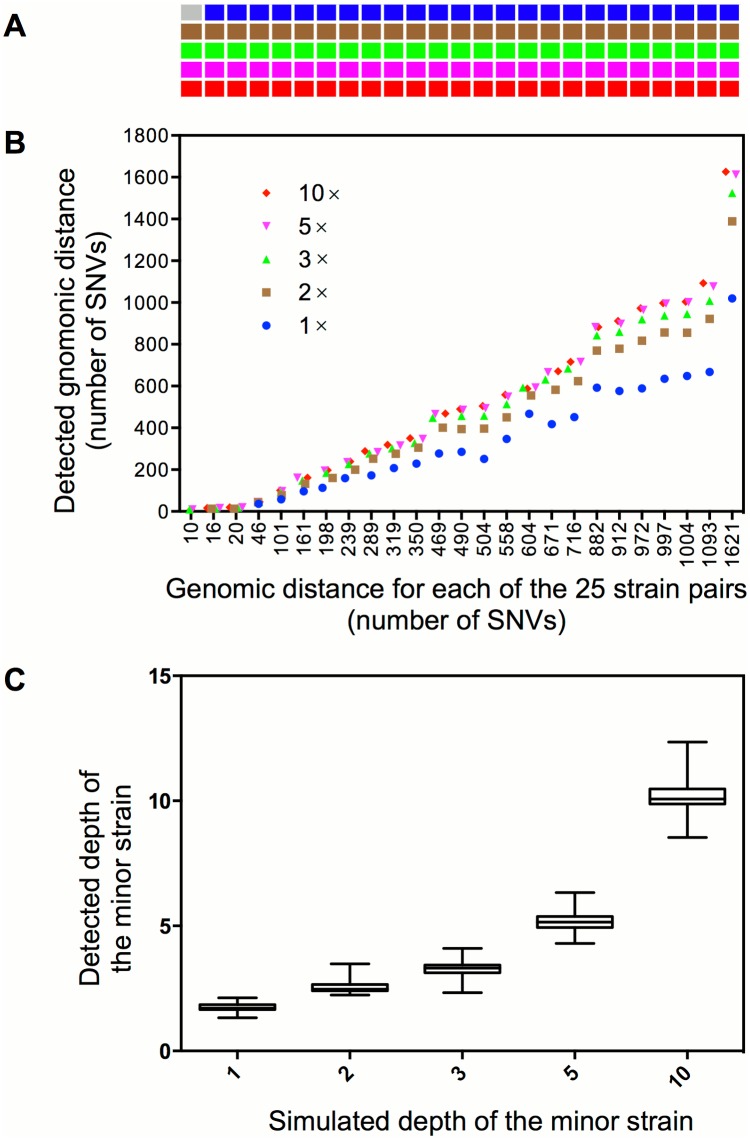
Mixed infection by multiple Mycobacterium tuberculosis (MTB) strains is associated with poor treatment outcome of tuberculosis (TB). Traditional genotyping methods have been used to detect mixed infections of MTB, however, their sensitivity and resolution are limited. Deep whole-genome sequencing (WGS) has been proved highly sensitive and discriminative for studying population heterogeneity of MTB. Here, we developed a phylogenetic-based method to detect MTB mixed infections using WGS data. We collected published WGS data of 782 global MTB strains from public database. We called homogeneous and heterogeneous single nucleotide variations (SNVs) of individual strains by mapping short reads to the ancestral MTB reference genome. We constructed a phylogenomic database based on 68,639 homogeneous SNVs of 652 MTB strains. Mixed infections were determined if multiple evolutionary paths were identified by mapping the SNVs of individual samples to the phylogenomic database. By simulation, our method could specifically detect mixed infections when the sequencing depth of minor strains was as low as 1× coverage, and when the genomic distance of two mixed strains was as small as 16 SNVs. By applying our methods to all 782 samples, we detected 47 mixed infections and 45 of them were caused by locally endemic strains. The results indicate that our method is highly sensitive and discriminative for identifying mixed infections from deep WGS data of MTB isolates.
Conflict of interest statement
Figures


References
-
- (WHO) WHO. Global tuberculosis report 2015. World Health Organization, Geneva, Switzerland 2015; Report WHO/HTM/TB/2015.22.
-
- Theisen A, Reichel C, Rusch-Gerdes S, Haas WH, Rockstroh JK, Spengler U, et al. Mixed-strain infection with a drug-sensitive and multidrug-resistant strain of Mycobacterium tuberculosis. Lancet. 1995;345(8963):1512 . - PubMed
-
- Chaves F, Dronda F, Alonso-Sanz M, Noriega AR. Evidence of exogenous reinfection and mixed infection with more than one strain of Mycobacterium tuberculosis among Spanish HIV-infected inmates. AIDS. 1999;13(5):615–20. Epub 1999/04/15. . - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
