

The genetic architecture of type 2 diabetes
- PMID: 27398621
- PMCID: PMC5034897
- DOI: 10.1038/nature18642
The genetic architecture of type 2 diabetes
Abstract
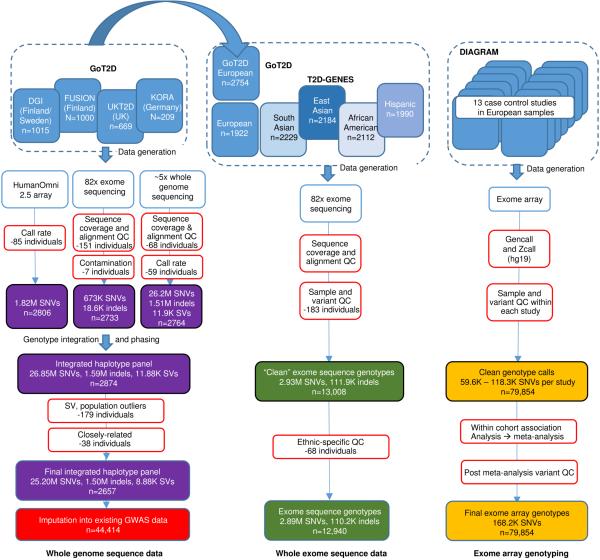
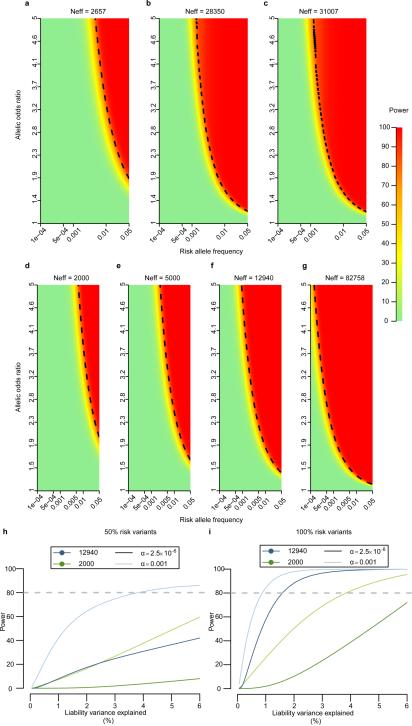
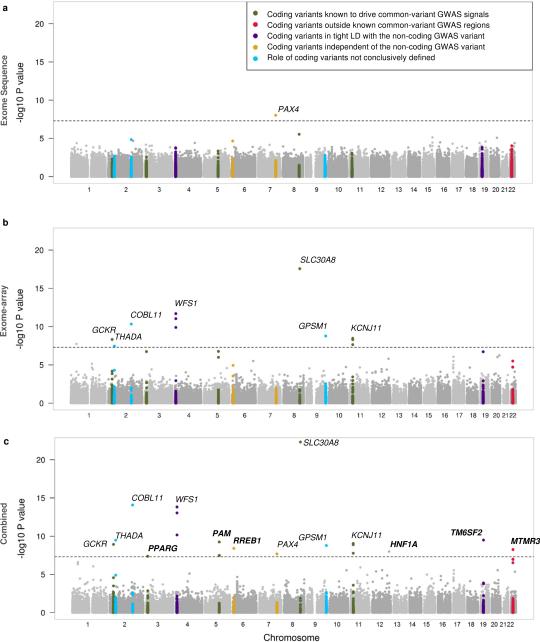
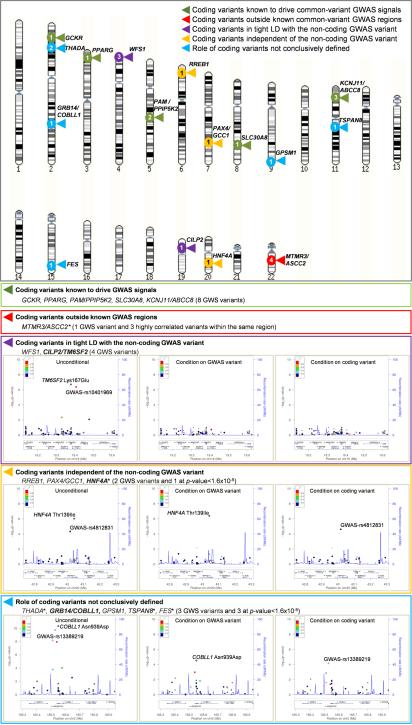
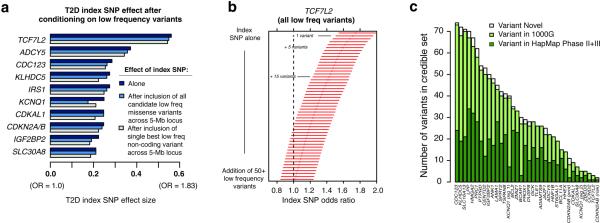
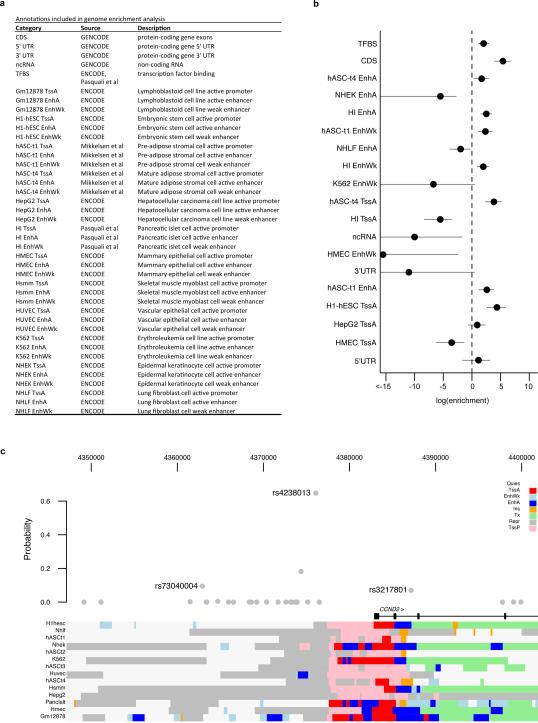
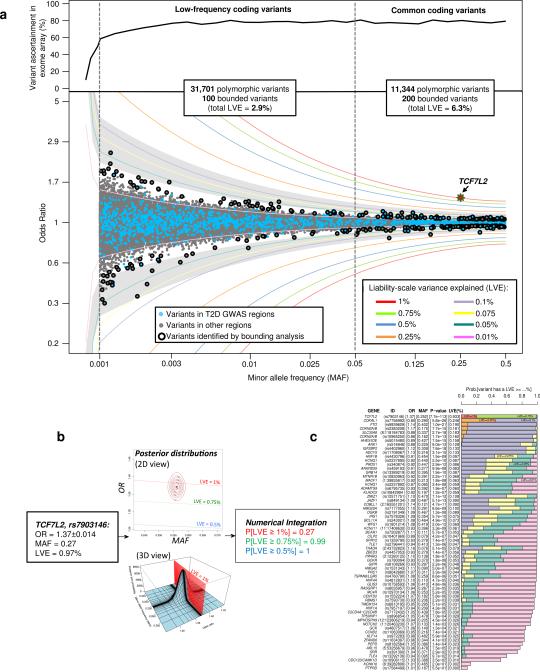
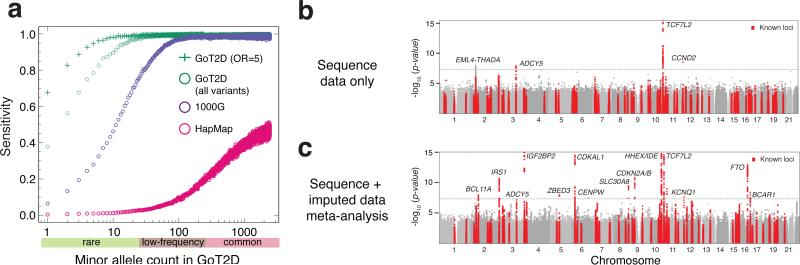
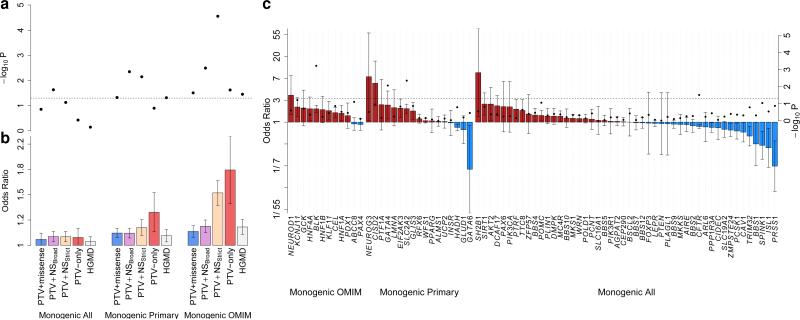
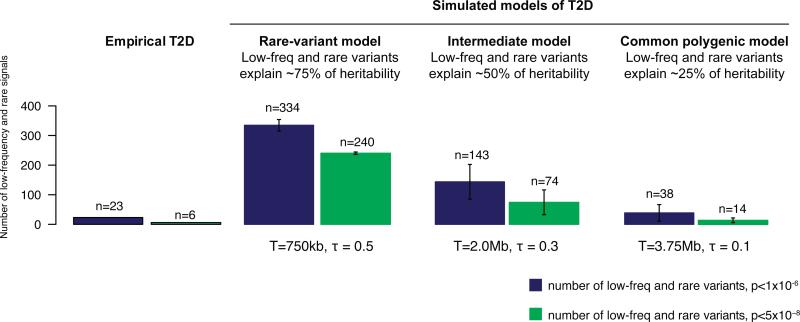
The genetic architecture of common traits, including the number, frequency, and effect sizes of inherited variants that contribute to individual risk, has been long debated. Genome-wide association studies have identified scores of common variants associated with type 2 diabetes, but in aggregate, these explain only a fraction of the heritability of this disease. Here, to test the hypothesis that lower-frequency variants explain much of the remainder, the GoT2D and T2D-GENES consortia performed whole-genome sequencing in 2,657 European individuals with and without diabetes, and exome sequencing in 12,940 individuals from five ancestry groups. To increase statistical power, we expanded the sample size via genotyping and imputation in a further 111,548 subjects. Variants associated with type 2 diabetes after sequencing were overwhelmingly common and most fell within regions previously identified by genome-wide association studies. Comprehensive enumeration of sequence variation is necessary to identify functional alleles that provide important clues to disease pathophysiology, but large-scale sequencing does not support the idea that lower-frequency variants have a major role in predisposition to type 2 diabetes.
Figures
Comment in
-
Diabetes: Still a geneticist's nightmare.Nature. 2016 Aug 4;536(7614):37-8. doi: 10.1038/nature18906. Epub 2016 Jul 13. Nature. 2016. PMID: 27398618 No abstract available.
References
-
- Willemsen G, et al. The concordance and heritability of type 2 diabetes in 34,166 Twin Pairs From International Twin Registers: The Discordant Twin (DISCOTWIN) Consortium. Twin Res Hum Genet. 2015;18:762–71. - PubMed
EXTENDED METHODS REFERENCES
-
- Guey LT, et al. Power in the phenotypic extremes: a simulation study of power in discovery and replication of rare variants. Genet Epidemiol. 2011;35:236–46. - PubMed
Publication types
MeSH terms
Grants and funding
- SP/04/002/BHF_/British Heart Foundation/United Kingdom
- P01AG027734/AG/NIA NIH HHS/United States
- R01 DK072193/DK/NIDDK NIH HHS/United States
- R01 HG000376/HG/NHGRI NIH HHS/United States
- 084723/WT_/Wellcome Trust/United Kingdom
- R01MH101820/MH/NIMH NIH HHS/United States
- R01AG046949/AG/NIA NIH HHS/United States
- R01 DK093757/DK/NIDDK NIH HHS/United States
- U01 DK085501/DK/NIDDK NIH HHS/United States
- U01 HG005773/HG/NHGRI NIH HHS/United States
- 083948/WT_/Wellcome Trust/United Kingdom
- R01 AG042188/AG/NIA NIH HHS/United States
- K12CA139160/CA/NCI NIH HHS/United States
- G0601966/MRC_/Medical Research Council/United Kingdom
- 098381/WT_/Wellcome Trust/United Kingdom
- K24 DK110550/DK/NIDDK NIH HHS/United States
- P60DK20595/DK/NIDDK NIH HHS/United States
- U01DK085501/DK/NIDDK NIH HHS/United States
- DK093757/DK/NIDDK NIH HHS/United States
- R00 DK099240/DK/NIDDK NIH HHS/United States
- 1RC2DK088389/DK/NIDDK NIH HHS/United States
- R00 DK092251/DK/NIDDK NIH HHS/United States
- 098017/WT_/Wellcome Trust/United Kingdom
- U54 MD007588/MD/NIMHD NIH HHS/United States
- R01 AG046949/AG/NIA NIH HHS/United States
- 090367/WT_/Wellcome Trust/United Kingdom
- MR/K002414/1/MRC_/Medical Research Council/United Kingdom
- U54 HG003067/HG/NHGRI NIH HHS/United States
- U01 DK085524/DK/NIDDK NIH HHS/United States
- K24DK080140/DK/NIDDK NIH HHS/United States
- DK085524/DK/NIDDK NIH HHS/United States
- R01DK066358/DK/NIDDK NIH HHS/United States
- U01 DK085545/DK/NIDDK NIH HHS/United States
- 095552/WT_/Wellcome Trust/United Kingdom
- R01 DK106236/DK/NIDDK NIH HHS/United States
- R01 DK098032/DK/NIDDK NIH HHS/United States
- U01DK085526/DK/NIDDK NIH HHS/United States
- U54HG003067/HG/NHGRI NIH HHS/United States
- 092447/WT_/Wellcome Trust/United Kingdom
- HHSN268201300048C/HL/NHLBI NIH HHS/United States
- T32 HG000040/HG/NHGRI NIH HHS/United States
- MC_UU_12012/5/MRC_/Medical Research Council/United Kingdom
- T32 GM007753/GM/NIGMS NIH HHS/United States
- R01MH090937/MH/NIMH NIH HHS/United States
- MR/L01341X/1/MRC_/Medical Research Council/United Kingdom
- R01DK062370/DK/NIDDK NIH HHS/United States
- S10 OD018522/OD/NIH HHS/United States
- R56 HG000376/HG/NHGRI NIH HHS/United States
- MC_UU_12015/1/MRC_/Medical Research Council/United Kingdom
- R01 DK101478/DK/NIDDK NIH HHS/United States
- R01 HL102830/HL/NHLBI NIH HHS/United States
- DK072193/DK/NIDDK NIH HHS/United States
- R01 DK073541/DK/NIDDK NIH HHS/United States
- P30AG038072/AG/NIA NIH HHS/United States
- R01 DK066358/DK/NIDDK NIH HHS/United States
- CIHR/Canada
- HHSN268201300049C/HL/NHLBI NIH HHS/United States
- 095101/WT_/Wellcome Trust/United Kingdom
- 100956/WT_/Wellcome Trust/United Kingdom
- P30DK020595/DK/NIDDK NIH HHS/United States
- K24 DK080140/DK/NIDDK NIH HHS/United States
- RC2 DK088389/DK/NIDDK NIH HHS/United States
- G0901213/MRC_/Medical Research Council/United Kingdom
- R01DK098032/DK/NIDDK NIH HHS/United States
- HHSN268201300047C/HL/NHLBI NIH HHS/United States
- 086596/WT_/Wellcome Trust/United Kingdom
- U01 DK085526/DK/NIDDK NIH HHS/United States
- RC2DK088389/DK/NIDDK NIH HHS/United States
- R00DK092251/DK/NIDDK NIH HHS/United States
- HHSN268201300050C/HL/NHLBI NIH HHS/United States
- U01 DK085584/DK/NIDDK NIH HHS/United States
- DK085584/DK/NIDDK NIH HHS/United States
- DK085545/DK/NIDDK NIH HHS/United States
- P01 AG027734/AG/NIA NIH HHS/United States
- U01 DK078616/DK/NIDDK NIH HHS/United States
- DK088389/DK/NIDDK NIH HHS/United States
- DK085526/DK/NIDDK NIH HHS/United States
- DK098032/DK/NIDDK NIH HHS/United States
- U01 DK062370/DK/NIDDK NIH HHS/United States
- P30 AG038072/AG/NIA NIH HHS/United States
- 090532/WT_/Wellcome Trust/United Kingdom
- 1R01AG042188/AG/NIA NIH HHS/United States
- DK085501/DK/NIDDK NIH HHS/United States
- 064890/WT_/Wellcome Trust/United Kingdom
- R01 MH101820/MH/NIMH NIH HHS/United States
- G0900747-‐91070/MRC_/Medical Research Council/United Kingdom
- U01HG005773/HG/NHGRI NIH HHS/United States
- P30 DK020572/DK/NIDDK NIH HHS/United States
- T32GM007753/GM/NIGMS NIH HHS/United States
- RG/14/5/30893/BHF_/British Heart Foundation/United Kingdom
- HHSN268201300046C/HL/NHLBI NIH HHS/United States
- 085475/WT_/Wellcome Trust/United Kingdom
- G0801566/MRC_/Medical Research Council/United Kingdom
- R01DK073541/DK/NIDDK NIH HHS/United States
- R01 MH090937/MH/NIMH NIH HHS/United States
- 200837/Z/16/Z/WT_/Wellcome Trust/United Kingdom
- MR/K013491/1/MRC_/Medical Research Council/United Kingdom
- T32 HL007055/HL/NHLBI NIH HHS/United States
- 098051/WT_/Wellcome Trust/United Kingdom
- RC2-‐DK088389/DK/NIDDK NIH HHS/United States
- G0601261/MRC_/Medical Research Council/United Kingdom
- SP/09/002/BHF_/British Heart Foundation/United Kingdom
- R01HL102830/HL/NHLBI NIH HHS/United States
- P30 DK020595/DK/NIDDK NIH HHS/United States
- G0800270/MRC_/Medical Research Council/United Kingdom
- P60 DK020595/DK/NIDDK NIH HHS/United States
- G0700931/MRC_/Medical Research Council/United Kingdom
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
