

From animal models to human disease: a genetic approach for personalized medicine in ALS
- PMID: 27400686
- PMCID: PMC4940869
- DOI: 10.1186/s40478-016-0340-5
From animal models to human disease: a genetic approach for personalized medicine in ALS
Abstract
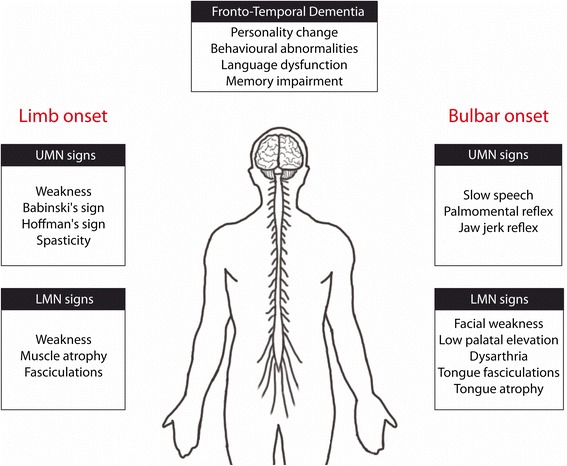
Amyotrophic Lateral Sclerosis (ALS) is the most frequent motor neuron disease in adults. Classical ALS is characterized by the death of upper and lower motor neurons leading to progressive paralysis. Approximately 10 % of ALS patients have familial form of the disease. Numerous different gene mutations have been found in familial cases of ALS, such as mutations in superoxide dismutase 1 (SOD1), TAR DNA-binding protein 43 (TDP-43), fused in sarcoma (FUS), C9ORF72, ubiquilin-2 (UBQLN2), optineurin (OPTN) and others. Multiple animal models were generated to mimic the disease and to test future treatments. However, no animal model fully replicates the spectrum of phenotypes in the human disease and it is difficult to assess how a therapeutic effect in disease models can predict efficacy in humans. Importantly, the genetic and phenotypic heterogeneity of ALS leads to a variety of responses to similar treatment regimens. From this has emerged the concept of personalized medicine (PM), which is a medical scheme that combines study of genetic, environmental and clinical diagnostic testing, including biomarkers, to individualized patient care. In this perspective, we used subgroups of specific ALS-linked gene mutations to go through existing animal models and to provide a comprehensive profile of the differences and similarities between animal models of disease and human disease. Finally, we reviewed application of biomarkers and gene therapies relevant in personalized medicine approach. For instance, this includes viral delivering of antisense oligonucleotide and small interfering RNA in SOD1, TDP-43 and C9orf72 mice models. Promising gene therapies raised possibilities for treating differently the major mutations in familial ALS cases.
Keywords: Amyotrophic lateral sclerosis (ALS); Animal models; Biomarkers; Frontotemporal dementia (FTD); Gene therapy; Mouse; Personalized medicine.
Figures



References
-
- Ajroud-Driss S, Siddique T. Sporadic and hereditary amyotrophic lateral sclerosis (ALS). Biochim Biophys Acta. 2014. doi:10.1016/j.bbadis.2014.08.010 - PubMed
-
- Ittner LM, Halliday GM, Kril JJ, Gotz J, Hodges JR, Kiernan MC. FTD and ALS--translating mouse studies into clinical trials. Nature reviews. Neurology. 2015;11(6):360–6. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Miscellaneous
