

The Nrd1-like protein Seb1 coordinates cotranscriptional 3' end processing and polyadenylation site selection
- PMID: 27401558
- PMCID: PMC4949328
- DOI: 10.1101/gad.280222.116
The Nrd1-like protein Seb1 coordinates cotranscriptional 3' end processing and polyadenylation site selection
Abstract
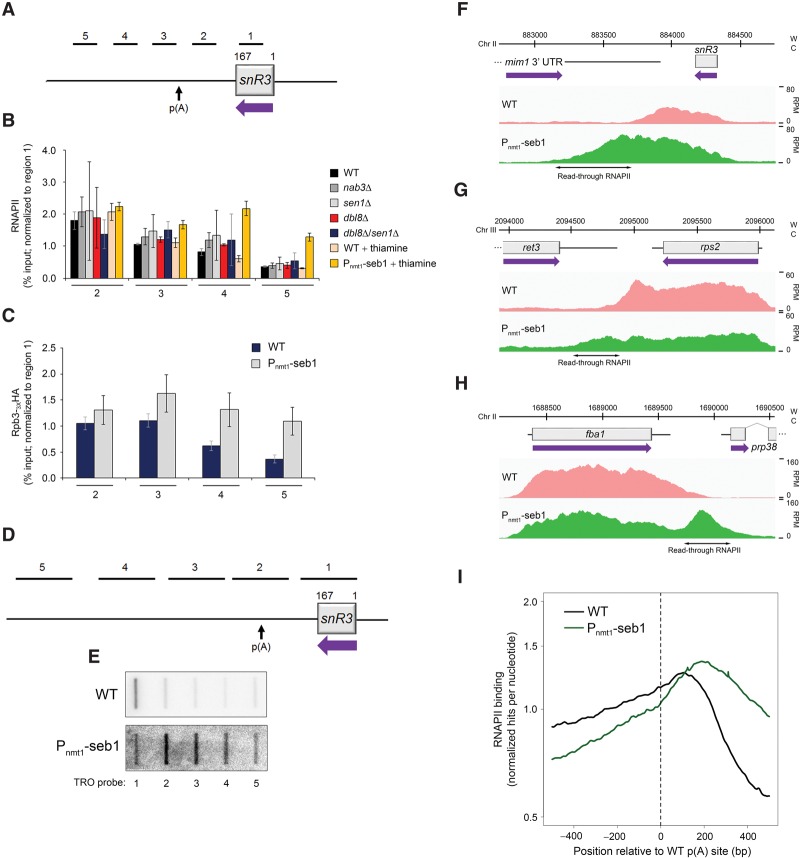
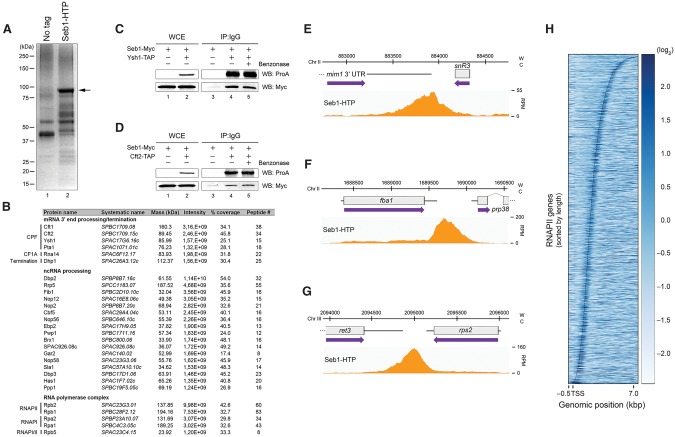
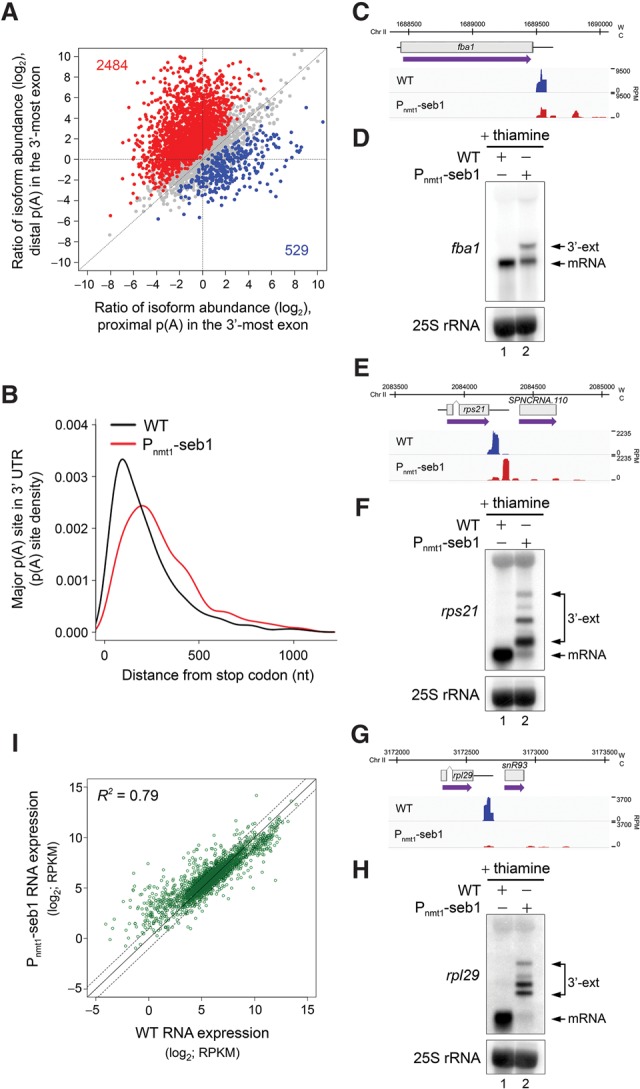
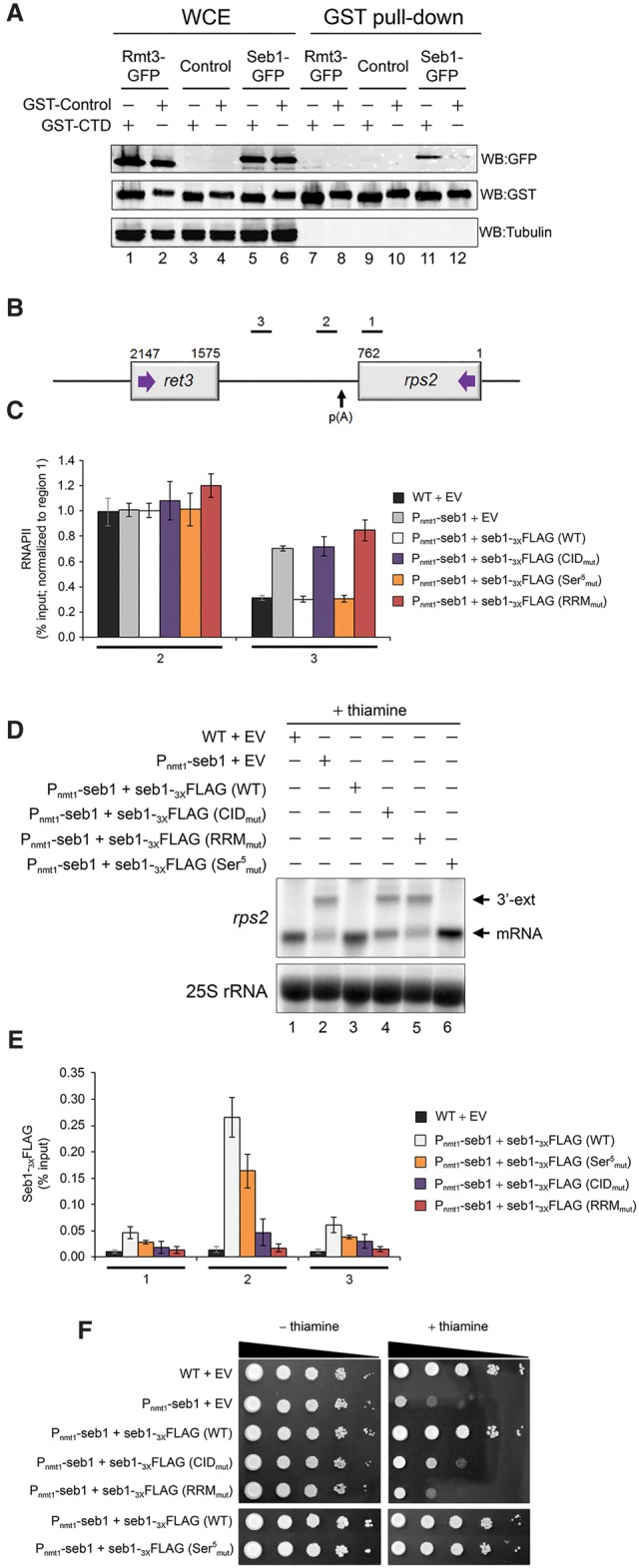
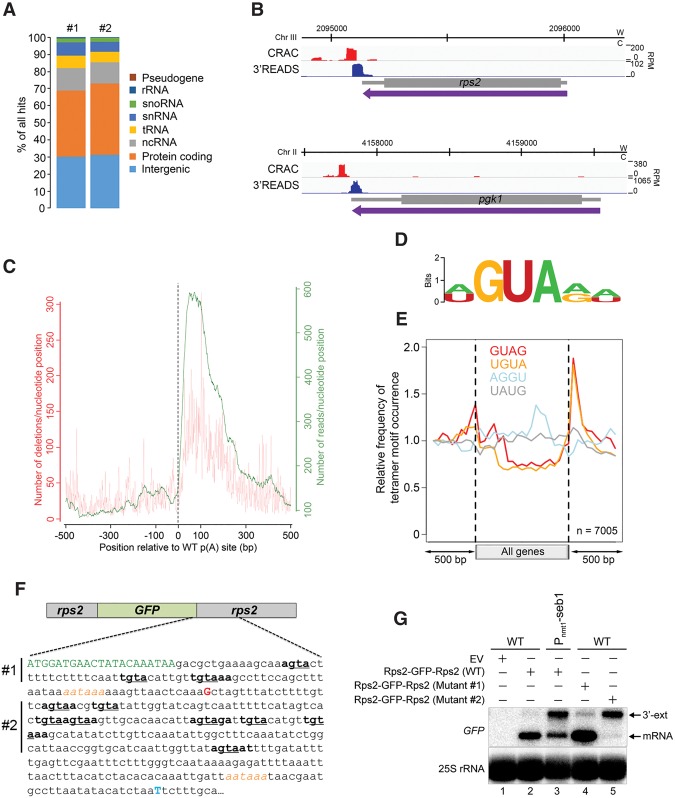
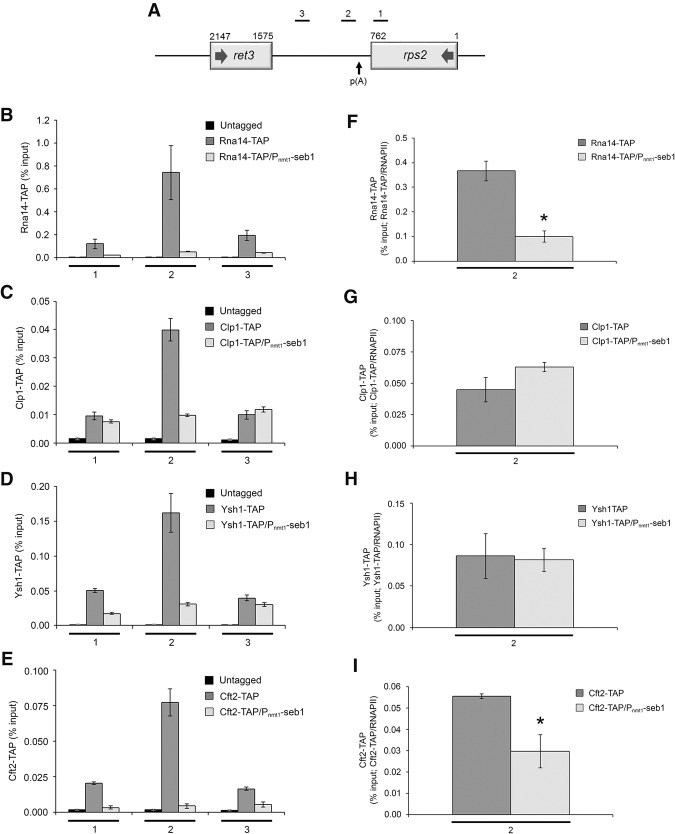
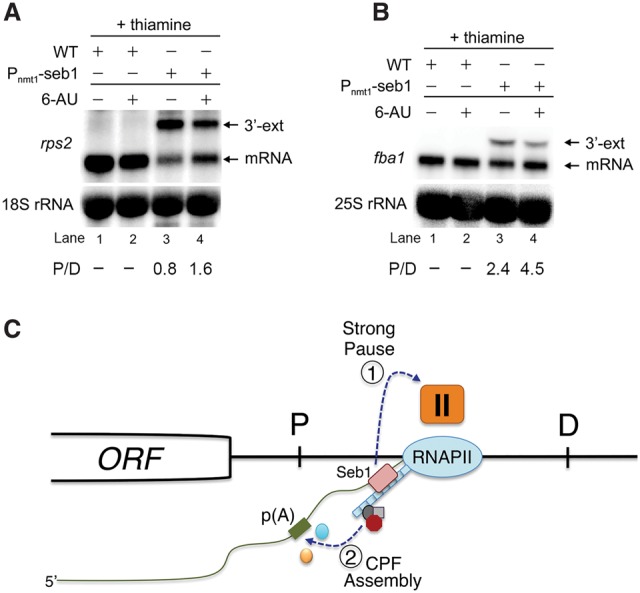
Termination of RNA polymerase II (RNAPII) transcription is associated with RNA 3' end formation. For coding genes, termination is initiated by the cleavage/polyadenylation machinery. In contrast, a majority of noncoding transcription events in Saccharomyces cerevisiae does not rely on RNA cleavage for termination but instead terminates via a pathway that requires the Nrd1-Nab3-Sen1 (NNS) complex. Here we show that the Schizosaccharomyces pombe ortholog of Nrd1, Seb1, does not function in NNS-like termination but promotes polyadenylation site selection of coding and noncoding genes. We found that Seb1 associates with 3' end processing factors, is enriched at the 3' end of genes, and binds RNA motifs downstream from cleavage sites. Importantly, a deficiency in Seb1 resulted in widespread changes in 3' untranslated region (UTR) length as a consequence of increased alternative polyadenylation. Given that Seb1 levels affected the recruitment of conserved 3' end processing factors, our findings indicate that the conserved RNA-binding protein Seb1 cotranscriptionally controls alternative polyadenylation.
Keywords: 3′ end processing; Nrd1; S. pombe; Seb1; polyadenylation; transcription termination.
© 2016 Lemay et al.; Published by Cold Spring Harbor Laboratory Press.
Figures
Similar articles
-
Polyadenylation site selection: linking transcription and RNA processing via a conserved carboxy-terminal domain (CTD)-interacting protein.Curr Genet. 2017 May;63(2):195-199. doi: 10.1007/s00294-016-0645-8. Epub 2016 Aug 31. Curr Genet. 2017. PMID: 27582274 Review.
-
RNA Polymerase II Transcription Attenuation at the Yeast DNA Repair Gene, DEF1, Involves Sen1-Dependent and Polyadenylation Site-Dependent Termination.G3 (Bethesda). 2018 May 31;8(6):2043-2058. doi: 10.1534/g3.118.200072. G3 (Bethesda). 2018. PMID: 29686108 Free PMC article.
-
Comparative analysis of alternative polyadenylation in S. cerevisiae and S. pombe.Genome Res. 2017 Oct;27(10):1685-1695. doi: 10.1101/gr.222331.117. Epub 2017 Sep 15. Genome Res. 2017. PMID: 28916539 Free PMC article.
-
Yeast Nrd1, Nab3, and Sen1 transcriptome-wide binding maps suggest multiple roles in post-transcriptional RNA processing.RNA. 2011 Nov;17(11):2011-25. doi: 10.1261/rna.2840711. Epub 2011 Sep 27. RNA. 2011. PMID: 21954178 Free PMC article.
-
The Nrd1-Nab3-Sen1 transcription termination complex from a structural perspective.Biochem Soc Trans. 2023 Jun 28;51(3):1257-1269. doi: 10.1042/BST20221418. Biochem Soc Trans. 2023. PMID: 37222282 Free PMC article. Review.
Cited by
-
Polyadenylated versions of small non-coding RNAs in Saccharomyces cerevisiae are degraded by Rrp6p/Rrp47p independent of the core nuclear exosome.Microb Cell. 2024 May 22;11:155-186. doi: 10.15698/mic2024.05.823. eCollection 2024. Microb Cell. 2024. PMID: 38783922 Free PMC article.
-
Bidirectional terminators in Saccharomyces cerevisiae prevent cryptic transcription from invading neighboring genes.Nucleic Acids Res. 2017 Jun 20;45(11):6417-6426. doi: 10.1093/nar/gkx242. Nucleic Acids Res. 2017. PMID: 28383698 Free PMC article.
-
Poly(A) site choice and Pol2 CTD Serine-5 status govern lncRNA control of phosphate-responsive tgp1 gene expression in fission yeast.RNA. 2018 Feb;24(2):237-250. doi: 10.1261/rna.063966.117. Epub 2017 Nov 9. RNA. 2018. PMID: 29122971 Free PMC article.
-
Autonomous Pathway: FLOWERING LOCUS C Repression through an Antisense-Mediated Chromatin-Silencing Mechanism.Plant Physiol. 2020 Jan;182(1):27-37. doi: 10.1104/pp.19.01009. Epub 2019 Nov 18. Plant Physiol. 2020. PMID: 31740502 Free PMC article.
-
Proteomic profiling and functional characterization of post-translational modifications of the fission yeast RNA exosome.Nucleic Acids Res. 2018 Nov 30;46(21):11169-11183. doi: 10.1093/nar/gky915. Nucleic Acids Res. 2018. PMID: 30321377 Free PMC article.
References
-
- Ahn SH, Kim M, Buratowski S. 2004. Phosphorylation of serine 2 within the RNA polymerase II C-terminal domain couples transcription and 3′ end processing. Mol Cell 13: 67–76. - PubMed
-
- Arigo JT, Eyler DE, Carroll KL, Corden JL. 2006. Termination of cryptic unstable transcripts is directed by yeast RNA-binding proteins Nrd1 and Nab3. Mol Cell 23: 841–851. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases