

GM2 gangliosidosis AB variant: novel mutation from India - a case report with a review
- PMID: 27402091
- PMCID: PMC4939586
- DOI: 10.1186/s12887-016-0626-6
GM2 gangliosidosis AB variant: novel mutation from India - a case report with a review
Abstract
Background: GM2 gangliosidosis-AB variants a rare autosomal recessive neurodegenerative disorder occurring due to deficiency of GM2 activator protein resulting from the mutation in GM2A gene. Only seven mutations in nine cases have been reported from different population except India.
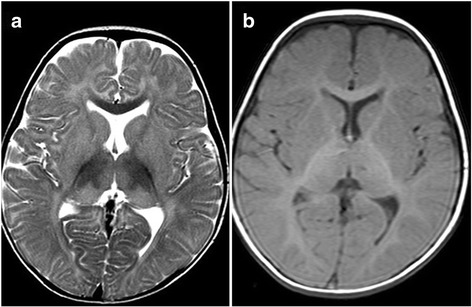
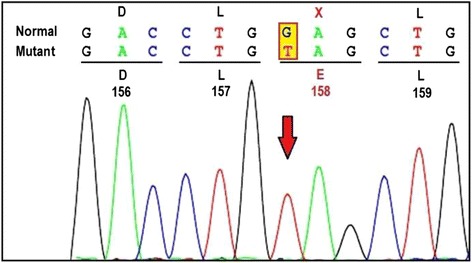
Case presentation: Present case is a one year old male born to 3rd degree consanguineous Indian parents from Maharashtra. He was presented with global developmental delay, hypotonia and sensitive to hyperacusis. Horizontal nystagmus and cherry red spot was detected during ophthalmic examination. MRI of brain revealed putaminal hyperintensity and thalamic hypointensity with some unmyelinated white matter in T2/T1 weighted images. Initially he was suspected having Tay-Sachs disease and finally diagnosed as GM2 gangliosidosis, AB variant due to truncated protein caused by nonsense mutation c.472 G > T (p.E158X) in GM2Agene.
Conclusion: Children with phenotypic presentation as GM2 gangliosidosis (Tay-Sachs or Sandhoff disease) and normal enzyme activity of β-hexosaminidase-A and -B in leucocytes need to be investigated for GM2 activator protein deficiency.
Keywords: AB variant; GM2 activator protein; GM2 gangliosidosis; GM2A gene.
Figures
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
