

Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer
- PMID: 27402485
- PMCID: PMC5256390
- DOI: 10.1136/gutjnl-2016-312078
Myeloid cells are required for PD-1/PD-L1 checkpoint activation and the establishment of an immunosuppressive environment in pancreatic cancer
Abstract
Background: Pancreatic cancer is characterised by the accumulation of a fibro-inflammatory stroma. Within this stromal reaction, myeloid cells are a predominant population. Distinct myeloid subsets have been correlated with tumour promotion and unmasking of anti-tumour immunity.
Objective: The goal of this study was to determine the effect of myeloid cell depletion on the onset and progression of pancreatic cancer and to understand the relationship between myeloid cells and T cell-mediated immunity within the pancreatic cancer microenvironment.
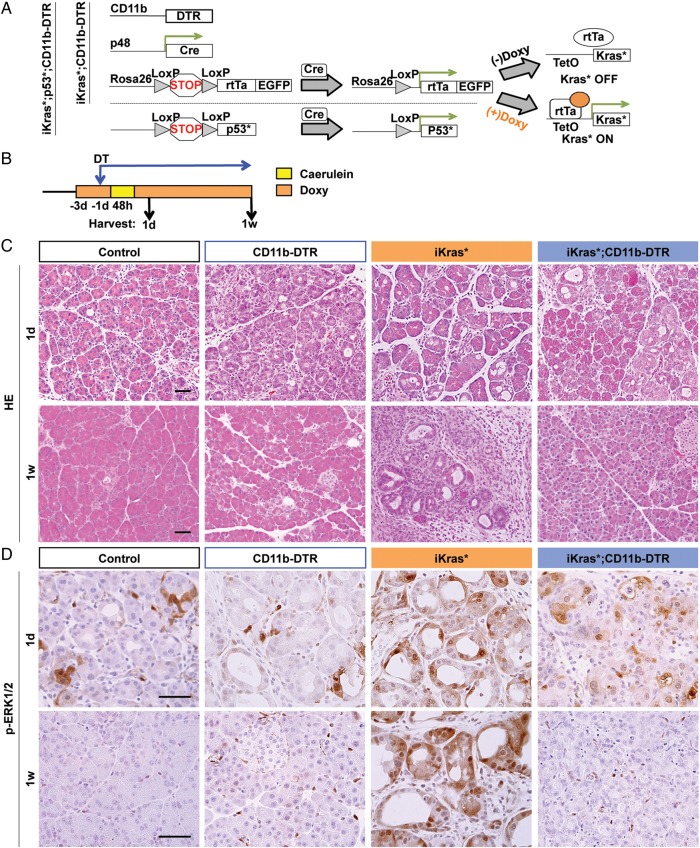
Methods: Primary mouse pancreatic cancer cells were transplanted into CD11b-diphtheria toxin receptor (DTR) mice. Alternatively, the iKras* mouse model of pancreatic cancer was crossed into CD11b-DTR mice. CD11b+ cells (mostly myeloid cell population) were depleted by diphtheria toxin treatment during tumour initiation or in established tumours.
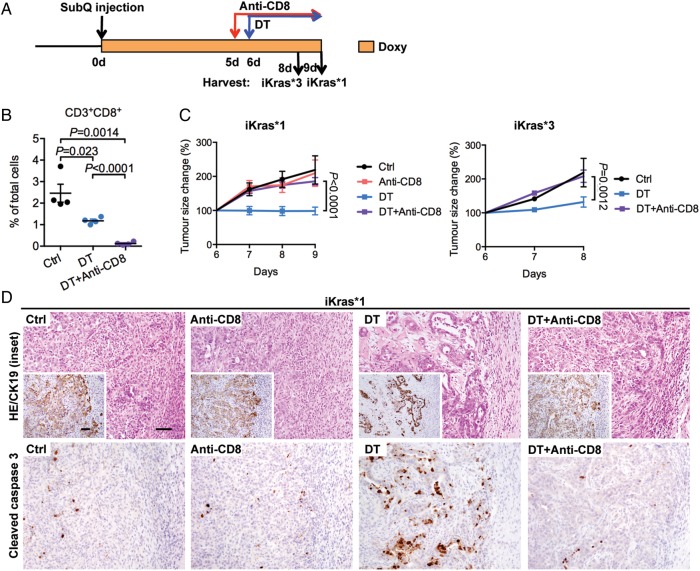
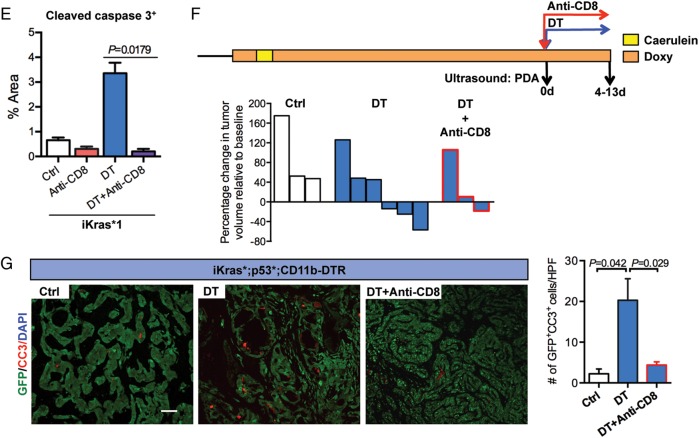
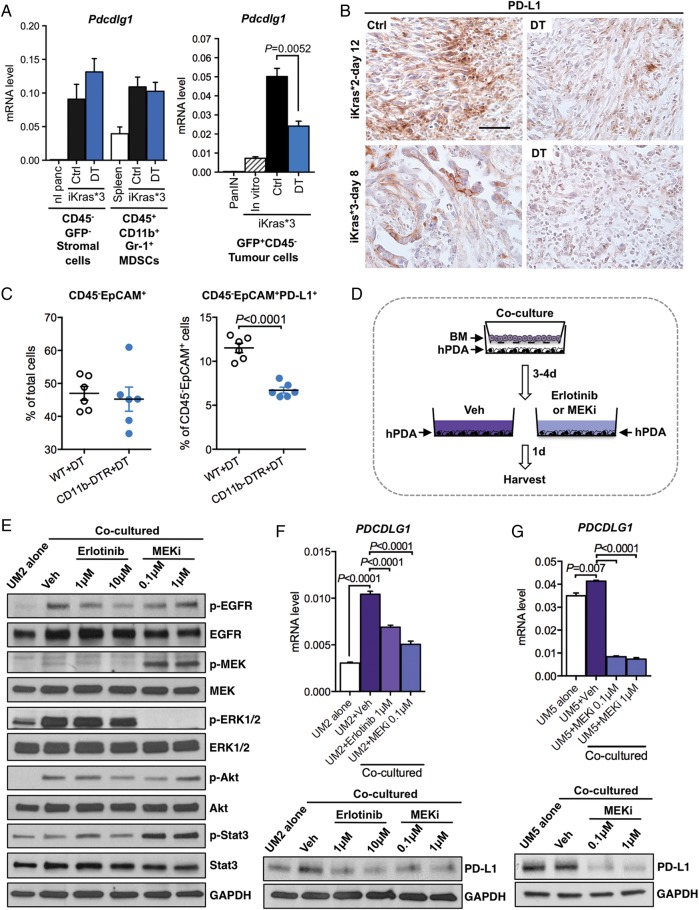
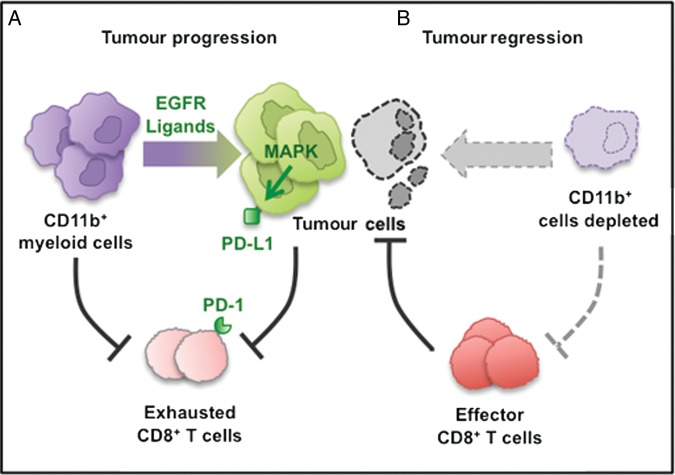
Results: Depletion of myeloid cells prevented KrasG12D-driven pancreatic cancer initiation. In pre-established tumours, myeloid cell depletion arrested tumour growth and in some cases, induced tumour regressions that were dependent on CD8+ T cells. We found that myeloid cells inhibited CD8+ T-cell anti-tumour activity by inducing the expression of programmed cell death-ligand 1 (PD-L1) in tumour cells in an epidermal growth factor receptor (EGFR)/mitogen-activated protein kinases (MAPK)-dependent manner.
Conclusion: Our results show that myeloid cells support immune evasion in pancreatic cancer through EGFR/MAPK-dependent regulation of PD-L1 expression on tumour cells. Derailing this crosstalk between myeloid cells and tumour cells is sufficient to restore anti-tumour immunity mediated by CD8+ T cells, a finding with implications for the design of immune therapies for pancreatic cancer.
Keywords: GENE REGULATION; IMMUNE RESPONSE; MACROPHAGES; PANCREATIC CANCER; SIGNAL TRANSDUCTION.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Conflict of interest statement
Conflicts of Interest: None declared.
Figures



Comment in
-
Refined strategies for the treatment of pancreatic carcinoma: targeting myeloid cells in order to overcome T cell exhaustion.Gut. 2017 Jan;66(1):3-4. doi: 10.1136/gutjnl-2016-312427. Epub 2016 Aug 31. Gut. 2017. PMID: 27582511 No abstract available.
-
Combined effect of anti-BAG3 and anti-PD-1 treatment on macrophage infiltrate, CD8+ T cell number and tumour growth in pancreatic cancer.Gut. 2018 Apr;67(4):780-782. doi: 10.1136/gutjnl-2017-314225. Epub 2017 Aug 11. Gut. 2018. PMID: 28801350 Free PMC article. No abstract available.
References
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Research Materials
Miscellaneous