

Controlling PTEN (Phosphatase and Tensin Homolog) Stability: A DOMINANT ROLE FOR LYSINE 66
- PMID: 27405757
- PMCID: PMC5000091
- DOI: 10.1074/jbc.M116.727750
Controlling PTEN (Phosphatase and Tensin Homolog) Stability: A DOMINANT ROLE FOR LYSINE 66
Abstract
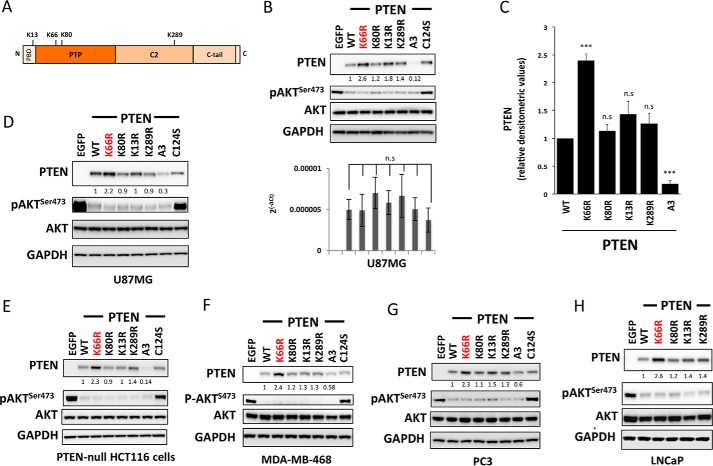
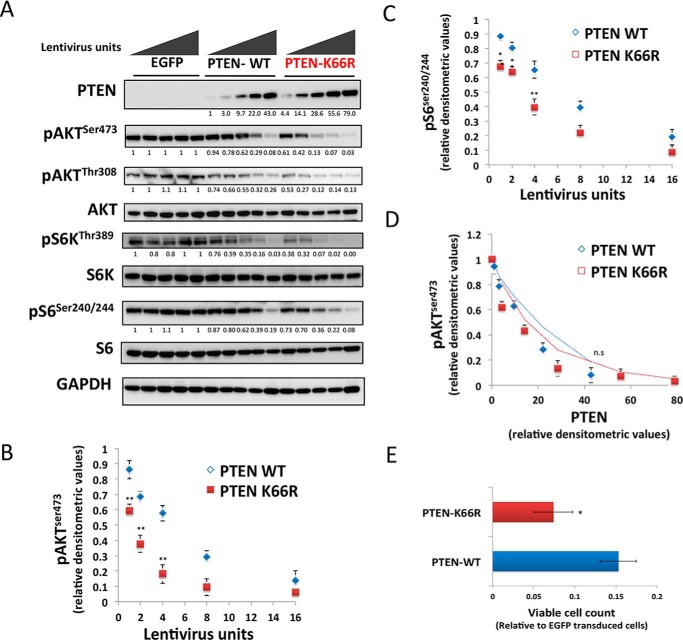
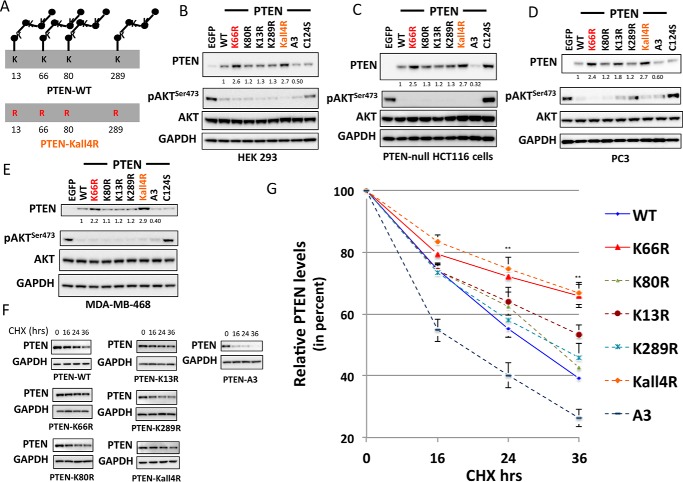
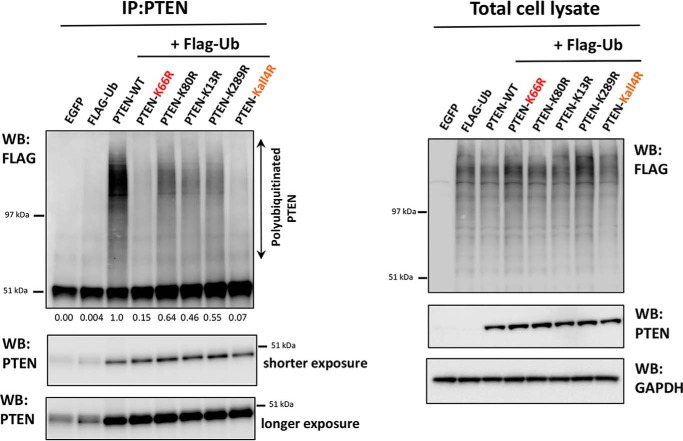
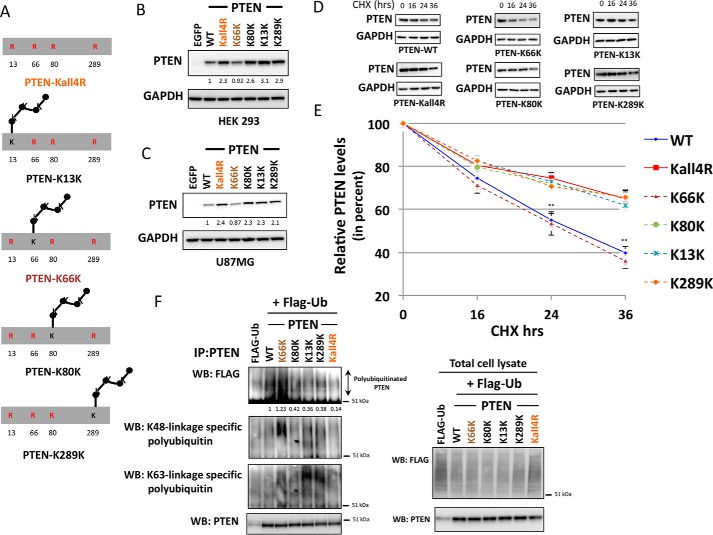
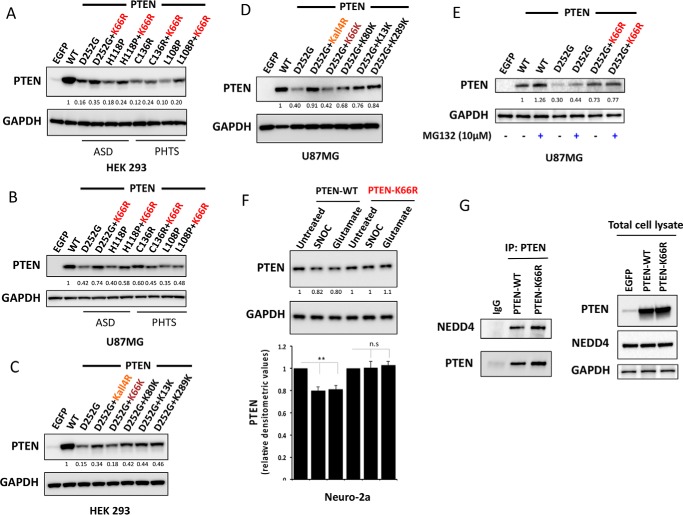
Phosphatase and tensin homolog (PTEN) is a phosphoinositide lipid phosphatase and one of the most frequently disrupted tumor suppressors in many forms of cancer, with even small reductions in the expression levels of PTEN promoting cancer development. Although the post-translational ubiquitination of PTEN can control its stability, activity, and localization, a detailed understanding of how PTEN ubiquitination integrates with other cellular regulatory processes and may be dysregulated in cancer has been hampered by a poor understanding of the significance of ubiquitination at individual sites. Here we show that Lys(66) is not required for cellular activity, yet dominates over other PTEN ubiquitination sites in the regulation of protein stability. Notably, combined mutation of other sites (Lys(13), Lys(80), and Lys(289)) has relatively little effect on protein expression, protein stability, or PTEN polyubiquitination. The present work identifies a key role for Lys(66) in the regulation of PTEN expression and provides both an opportunity to improve the stability of PTEN as a protein therapy and a mechanistic basis for efforts to stabilize endogenous PTEN.
Keywords: cancer; phosphatase; phosphatase and tensin homolog (PTEN); phosphoinositide; phosphoinositide 3-kinase; protein stability; tumor suppressor; ubiquitylation (ubiquitination).
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
Similar articles
-
Ubiquitination of PTEN (phosphatase and tensin homolog) inhibits phosphatase activity and is enhanced by membrane targeting and hyperosmotic stress.J Biol Chem. 2010 Apr 23;285(17):12620-8. doi: 10.1074/jbc.M109.072280. Epub 2010 Feb 22. J Biol Chem. 2010. PMID: 20177066 Free PMC article.
-
Characterization of PTEN mutations in brain cancer reveals that pten mono-ubiquitination promotes protein stability and nuclear localization.Oncogene. 2017 Jun 29;36(26):3673-3685. doi: 10.1038/onc.2016.493. Epub 2017 Mar 6. Oncogene. 2017. PMID: 28263967 Free PMC article.
-
Analysis of PTEN ubiquitylation and SUMOylation using molecular traps.Methods. 2015 May;77-78:112-8. doi: 10.1016/j.ymeth.2014.09.001. Epub 2014 Sep 16. Methods. 2015. PMID: 25224693
-
Ubiquitination/de-ubiquitination: A promising therapeutic target for PTEN reactivation in cancer.Biochim Biophys Acta Rev Cancer. 2022 May;1877(3):188723. doi: 10.1016/j.bbcan.2022.188723. Epub 2022 Mar 18. Biochim Biophys Acta Rev Cancer. 2022. PMID: 35314212 Review.
-
Post-translational regulation of PTEN.Oncogene. 2008 Sep 18;27(41):5454-63. doi: 10.1038/onc.2008.242. Oncogene. 2008. PMID: 18794880 Review.
Cited by
-
Topical MTII Therapy Suppresses Melanoma Through PTEN Upregulation and Cyclooxygenase II Inhibition.Int J Mol Sci. 2020 Jan 20;21(2):681. doi: 10.3390/ijms21020681. Int J Mol Sci. 2020. PMID: 31968661 Free PMC article.
-
Cowden syndrome-associated germline succinate dehydrogenase complex subunit D (SDHD) variants cause PTEN-mediated down-regulation of autophagy in thyroid cancer cells.Hum Mol Genet. 2017 Apr 1;26(7):1365-1375. doi: 10.1093/hmg/ddx037. Hum Mol Genet. 2017. PMID: 28164237 Free PMC article.
-
Colonic Inhibition of Phosphatase and Tensin Homolog Increases Colitogenic Bacteria, Causing Development of Colitis in Il10-/- Mice.Inflamm Bowel Dis. 2018 Jul 12;24(8):1718-1732. doi: 10.1093/ibd/izy124. Inflamm Bowel Dis. 2018. PMID: 29788382 Free PMC article.
-
The functions of tumor suppressor PTEN in innate and adaptive immunity.Cell Mol Immunol. 2017 Jul;14(7):581-589. doi: 10.1038/cmi.2017.30. Epub 2017 Jun 26. Cell Mol Immunol. 2017. PMID: 28603282 Free PMC article. Review.
-
Precise Immunodetection of PTEN Protein in Human Neoplasia.Cold Spring Harb Perspect Med. 2019 Dec 2;9(12):a036293. doi: 10.1101/cshperspect.a036293. Cold Spring Harb Perspect Med. 2019. PMID: 31501265 Free PMC article. Review.
References
-
- Leslie N. R., and Foti M. (2011) Non-genomic loss of PTEN function in cancer: not in my genes. Trends Pharmacol. Sci. 32, 131–140 - PubMed
-
- Song M. S., Salmena L., and Pandolfi P. P. (2012) The functions and regulation of the PTEN tumour suppressor. Nat. Rev. Mol. Cell Biol. 13, 283–296 - PubMed
-
- Worby C. A., and Dixon J. E. (2014) PTEN. Annu. Rev. Biochem. 83, 641–669 - PubMed
-
- Hopkins B. D., Fine B., Steinbach N., Dendy M., Rapp Z., Shaw J., Pappas K., Yu J. S., Hodakoski C., Mense S., Klein J., Pegno S., Sulis M. L., Goldstein H., Amendolara B., et al. (2013) A secreted PTEN phosphatase that enters cells to alter signaling and survival. Science 341, 399–402 - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
