

Mobile genes in the human microbiome are structured from global to individual scales
- PMID: 27409808
- PMCID: PMC4983458
- DOI: 10.1038/nature18927
Mobile genes in the human microbiome are structured from global to individual scales
Erratum in
-
Corrigendum: Mobile genes in the human microbiome are structured from global to individual scales.Nature. 2017 Apr 6;544(7648):124. doi: 10.1038/nature20774. Epub 2017 Mar 22. Nature. 2017. PMID: 28329759 No abstract available.
Abstract
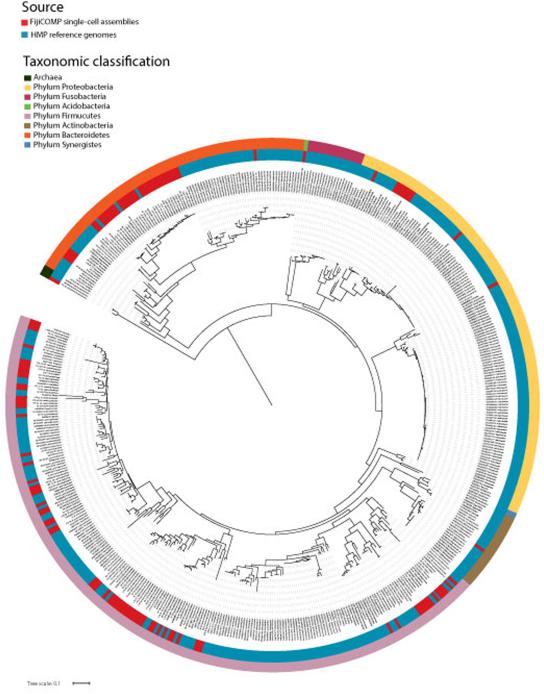
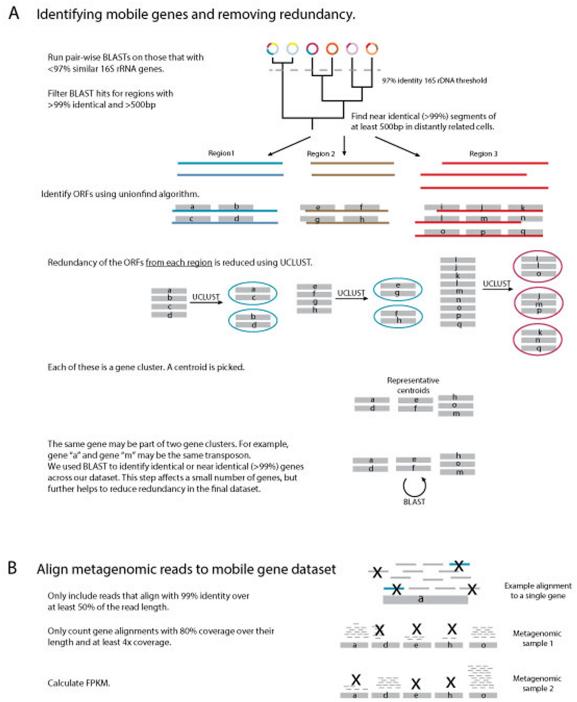
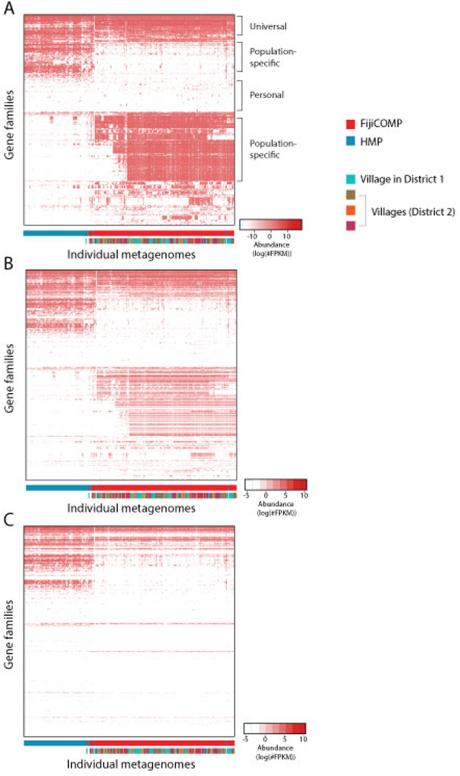
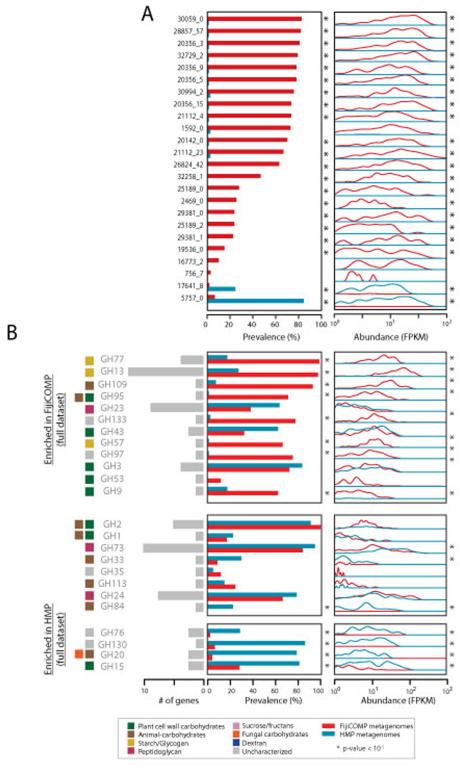
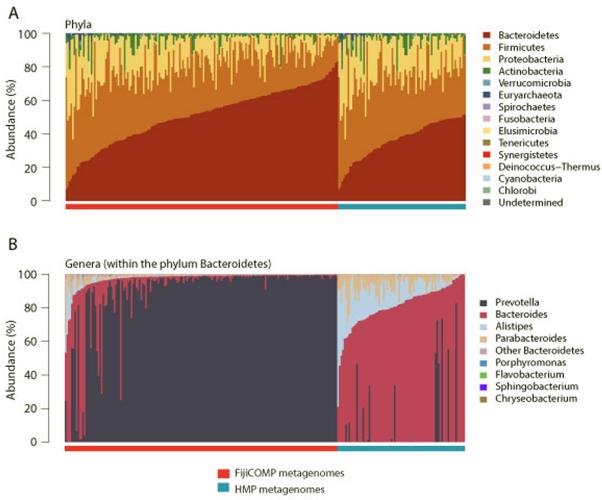
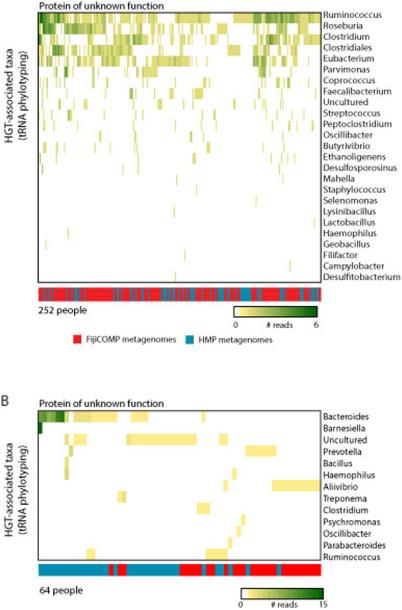
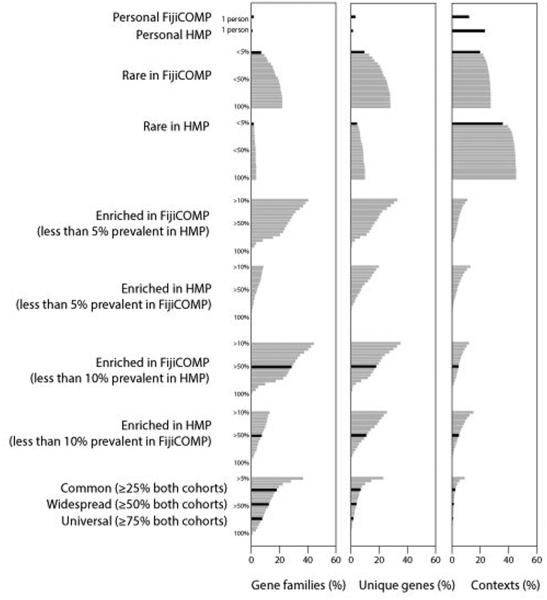
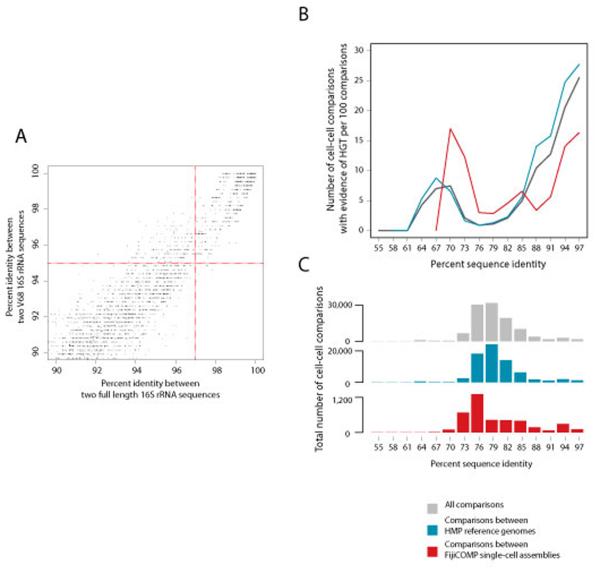
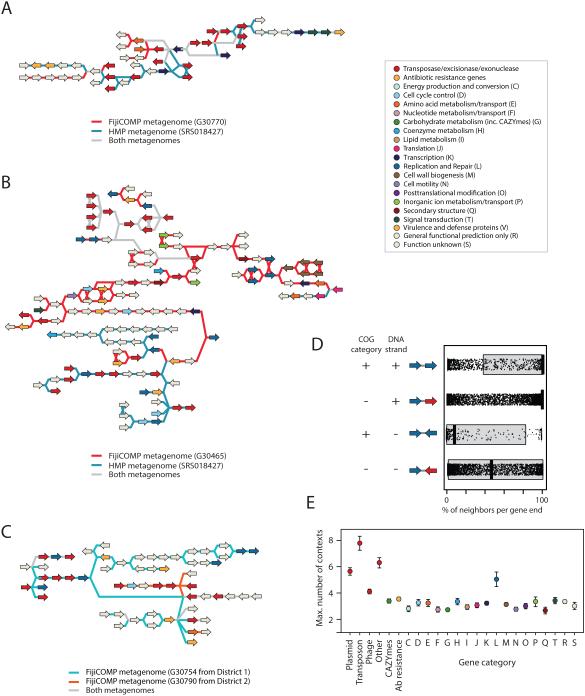
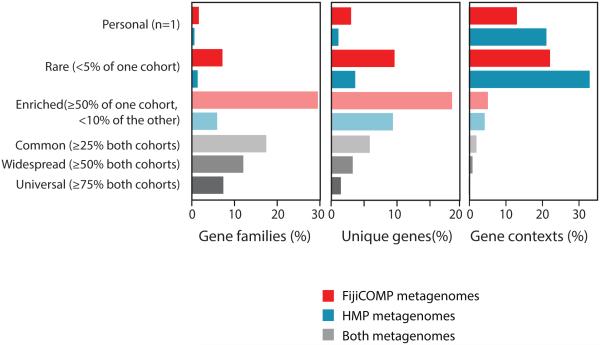
Recent work has underscored the importance of the microbiome in human health, and has largely attributed differences in phenotype to differences in the species present among individuals. However, mobile genes can confer profoundly different phenotypes on different strains of the same species. Little is known about the function and distribution of mobile genes in the human microbiome, and in particular whether the gene pool is globally homogenous or constrained by human population structure. Here, we investigate this question by comparing the mobile genes found in the microbiomes of 81 metropolitan North Americans with those of 172 agrarian Fiji islanders using a combination of single-cell genomics and metagenomics. We find large differences in mobile gene content between the Fijian and North American microbiomes, with functional variation that mirrors known dietary differences such as the excess of plant-based starch degradation genes found in Fijian individuals. Notably, we also observed differences between the mobile gene pools of neighbouring Fijian villages, even though microbiome composition across villages is similar. Finally, we observe high rates of recombination leading to individual-specific mobile elements, suggesting that the abundance of some genes may reflect environmental selection rather than dispersal limitation. Together, these data support the hypothesis that human activities and behaviours provide selective pressures that shape mobile gene pools, and that acquisition of mobile genes is important for colonizing specific human populations.
Figures
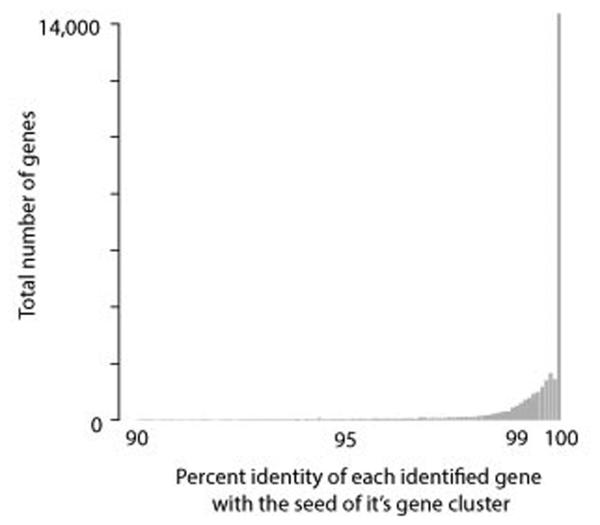
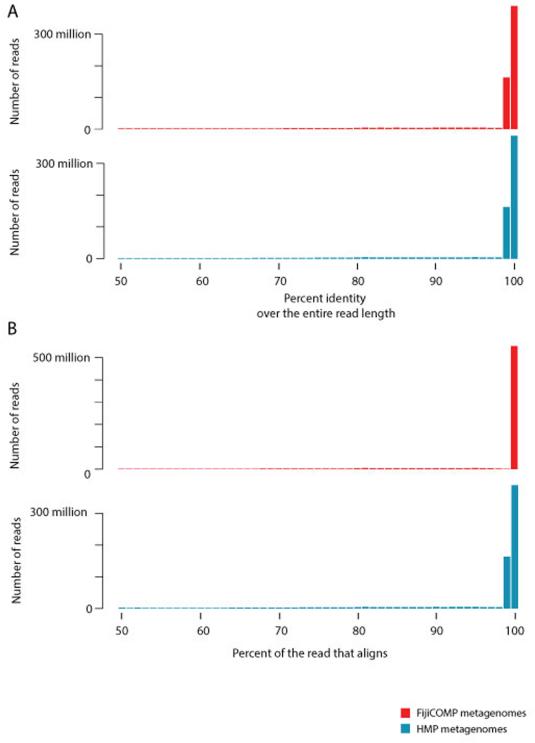
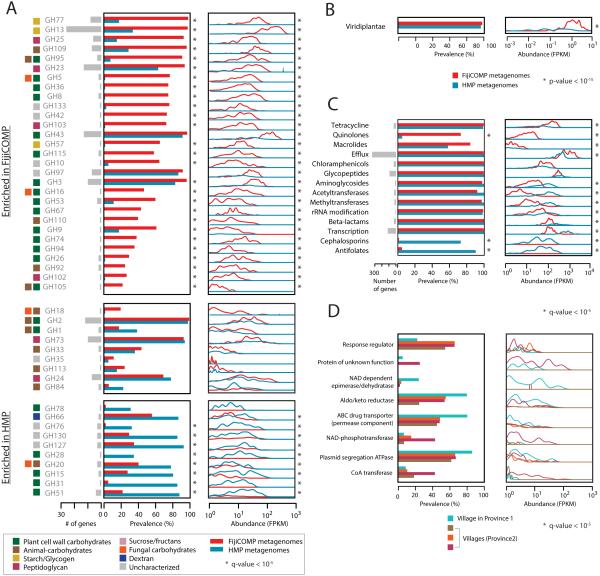
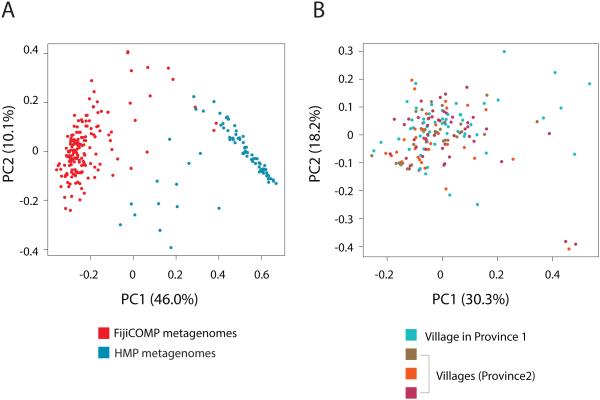
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
