

Nerve conduction velocity in CMT1A: what else can we tell?
- PMID: 27412484
- PMCID: PMC5603914
- DOI: 10.1111/ene.13079
Nerve conduction velocity in CMT1A: what else can we tell?
Abstract
Background and purpose: Charcot-Marie-Tooth disease (CMT) type 1A is characterized by uniformly reduced nerve conduction velocity (NCV) that is fully penetrant since the first years of life, remains fairly stable through the life and does not correlate with disability whereas compound muscular action potential (CMAP) amplitude does. The aim of the present study was to analyze the large amount of electrophysiological data collected in the ascorbic acid trial in Italy and the UK (CMT-TRIAAL/CMT-TRAUK) and to use these data to gain insights into the pathophysiology of NCV in CMT1A.
Methods: Baseline electrophysiological data from 271 patients were analysed. Electrophysiological recordings were taken from the motor ulnar, median and peroneal nerves and the sensory ulnar nerve. Distal motor latency (DML), motor (MNCV) and sensory (SNCV) nerve conduction velocity, and amplitudes of CMAPs and sensory action potentials were assessed. Electrophysiological findings were correlated with age of patients at examination and the Charcot-Marie-Tooth Examination Score (CMTES).
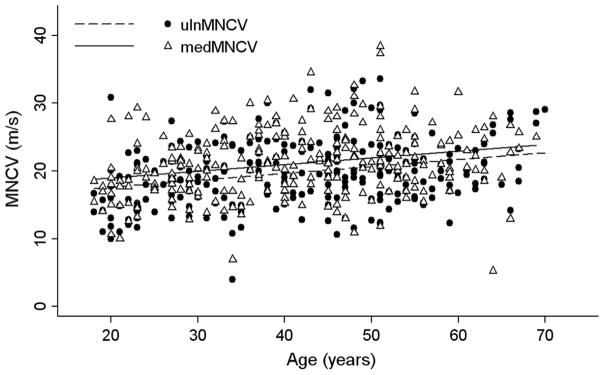
Results: NCV was markedly and uniformly reduced. CMAP amplitudes were overall reduced but more severely in lower limbs. DML decreased and MNCV and SNCV increased with age of the patients, whereas CMAP amplitudes worsened with age and also correlated with CMTES.
Conclusions: This is the largest sample of electrophysiological data obtained so far from CMT1A patients. Axonal degeneration as assessed by means of CMAP amplitude reflected clinical impairment and was consistent with a slowly progressive length-dependent neuropathy. All patients typically had markedly slowed NCV that did, however, slightly increase with age of the patients. The improvement of NCV might depend on myelin thickness remodelling that occurs during the adult life of CMT1A patients.
Keywords: CMT-TRIAAL/CMT-TRAUK; CMT1A; Charcot−Marie−Tooth disease; hereditary neuropathies; nerve conduction velocity.
© 2016 EAN.
Conflict of interest statement
Dr Solari reports grants from the Foundation of the Italian National Multiple Sclerosis Society, was a board member for Biogen Idec, Novartis, and has received speaker honoraria from Biogen Idec, Novartis, Excemed, Genzyme, Merck Serono and Teva. All other authors declare that they have no conflicts of interest.
Figures
References
-
- Shy M, Lupski JR, Chance PF, Klein CJ, Dyck PJ. Hereditary motor and sensory neuropathies: an overview of clinical, genetic, electrophysiologic and pathologic features. In: Ravi A, editor. Peripheral Neuropathy. 4. Philadelphia: Elsevier Saunders; 2005. pp. 1623–1658.
-
- Birouk N, Gouider R, Le Guern E, et al. Charcot–Marie–Tooth disease type 1A with 17p11.2 duplication. Clinical and electrophysiological phenotype study and factors influencing disease severity in 119 cases. Brain. 1997;120:813–823. - PubMed
-
- Krajewski KM, Lewis RA, Fuerst DR, et al. Neurological dysfunction and axonal degeneration in Charcot–Marie–Tooth disease type 1A. Brain. 2000;123:1516–1527. - PubMed
-
- García A, Combarros O, Calleja J, Berciano J. Charcot–Marie–Tooth disease type 1A with 17p duplication in infancy and early childhood: a longitudinal clinical and electrophysiologic study. Neurology. 1998;50:1061–1067. - PubMed
-
- Berciano J, García A, Calleja J, Combarros O. Clinico-electrophysiological correlation of extensor digitorum brevis muscle atrophy in children with Charcot–Marie–Tooth disease 1A duplication. Neuromuscul Disord. 2000;10:419–424. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
