

The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease
- PMID: 27412952
- PMCID: PMC5264221
- DOI: 10.1136/jmedgenet-2016-103910
The clinical, biochemical and genetic features associated with RMND1-related mitochondrial disease
Abstract
Background: Mutations in the RMND1 (Required for Meiotic Nuclear Division protein 1) gene have recently been linked to infantile onset mitochondrial disease characterised by multiple mitochondrial respiratory chain defects.
Methods: We summarised the clinical, biochemical and molecular genetic investigation of an international cohort of affected individuals with RMND1 mutations. In addition, we reviewed all the previously published cases to determine the genotype-phenotype correlates and performed survival analysis to identify prognostic factors.
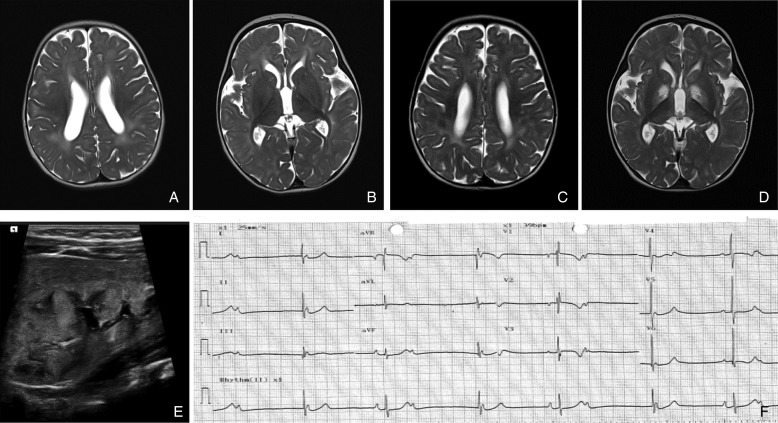
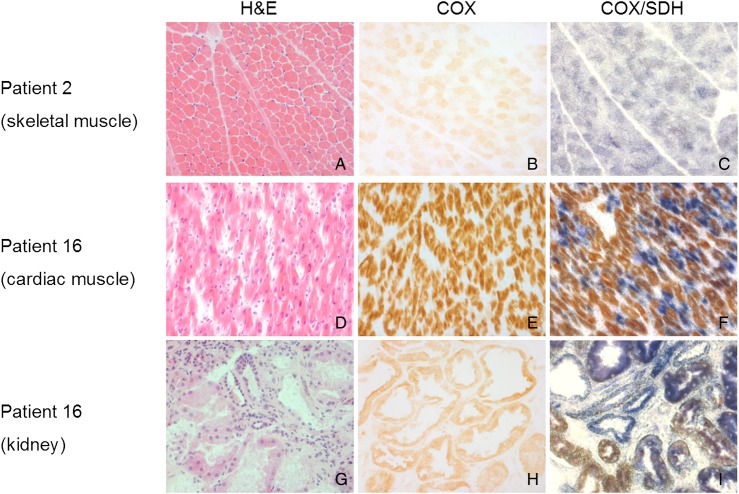
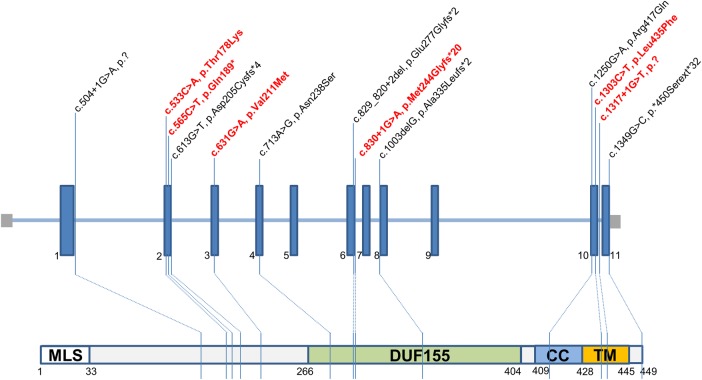
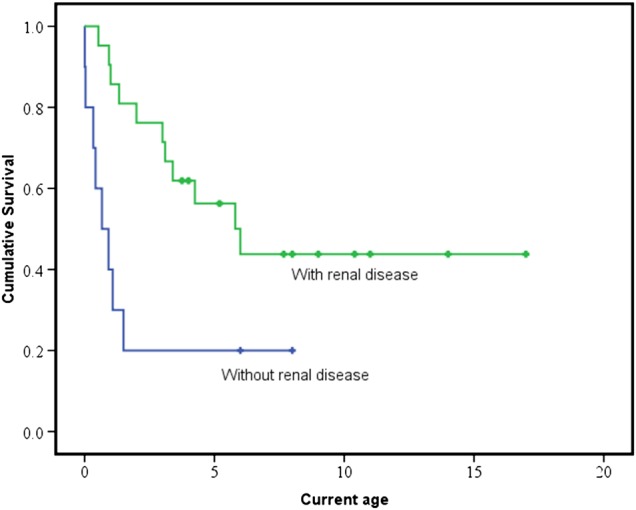
Results: We identified 14 new cases from 11 pedigrees that harbour recessive RMND1 mutations, including 6 novel variants: c.533C>A, p.(Thr178Lys); c.565C>T, p.(Gln189*); c.631G>A, p.(Val211Met); c.1303C>T, p.(Leu435Phe); c.830+1G>A and c.1317+1G>T. Together with all previously published cases (n=32), we show that congenital sensorineural deafness, hypotonia, developmental delay and lactic acidaemia are common clinical manifestations with disease onset under 2 years. Renal involvement is more prevalent than seizures (66% vs 44%). In addition, median survival time was longer in patients with renal involvement compared with those without renal disease (6 years vs 8 months, p=0.009). The neurological phenotype also appears milder in patients with renal involvement.
Conclusions: The clinical phenotypes and prognosis associated with RMND1 mutations are more heterogeneous than that were initially described. Regular monitoring of kidney function is imperative in the clinical practice in light of nephropathy being present in over 60% of cases. Furthermore, renal replacement therapy should be considered particularly in those patients with mild neurological manifestation as shown in our study that four recipients of kidney transplant demonstrate good clinical outcome to date.
Keywords: congenital sensorineural deafness; lactic acidosis; mitochondrial respiratory chain deficiencies; prognosis; renal disease.
Published by the BMJ Publishing Group Limited. For permission to use (where not already granted under a licence) please go to http://www.bmj.com/company/products-services/rights-and-licensing/.
Conflict of interest statement
Competing interests: None declared.
Figures
References
-
- Garcia-Diaz B, Barros MH, Sanna-Cherchi S, Emmanuele V, Akman HO, Ferreiro-Barros CC, Horvath R, Tadesse S, El Gharaby N, DiMauro S, De Vivo DC, Shokr A, Hirano M, Quinzii CM. Infantile encephaloneuromyopathy and defective mitochondrial translation are due to a homozygous RMND1 mutation. Am J Hum Genet 2012;91:729–36. 10.1016/j.ajhg.2012.08.019 - DOI - PMC - PubMed
-
- Janer A, Antonicka H, Lalonde E, Nishimura T, Sasarman F, Brown GK, Brown RM, Majewski J, Shoubridge EA. An RMND1 Mutation causes encephalopathy associated with multiple oxidative phosphorylation complex deficiencies and a mitochondrial translation defect. Am J Hum Genet 2012;91:737–43. 10.1016/j.ajhg.2012.08.020 - DOI - PMC - PubMed
-
- Janer A, van Karnebeek CD, Sasarman F, Antonicka H, Al Ghamdi M, Shyr C, Dunbar M, Stockler-Ispiroglu S, Ross CJ, Vallance H, Dionne J, Wasserman WW, Shoubridge EA. RMND1 deficiency associated with neonatal lactic acidosis, infantile onset renal failure, deafness, and multiorgan involvement. Eur J Hum Genet 2015;23:1301–7. 10.1038/ejhg.2014.293 - DOI - PMC - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials