

Sequence CLCN1 and SCN4A in patients with Nondystrophic myotonias in Chinese populations: Genetic and pedigree analysis of 10 families and review of the literature
- PMID: 27415035
- PMCID: PMC5279883
- DOI: 10.1080/19336950.2016.1212140
Sequence CLCN1 and SCN4A in patients with Nondystrophic myotonias in Chinese populations: Genetic and pedigree analysis of 10 families and review of the literature
Abstract
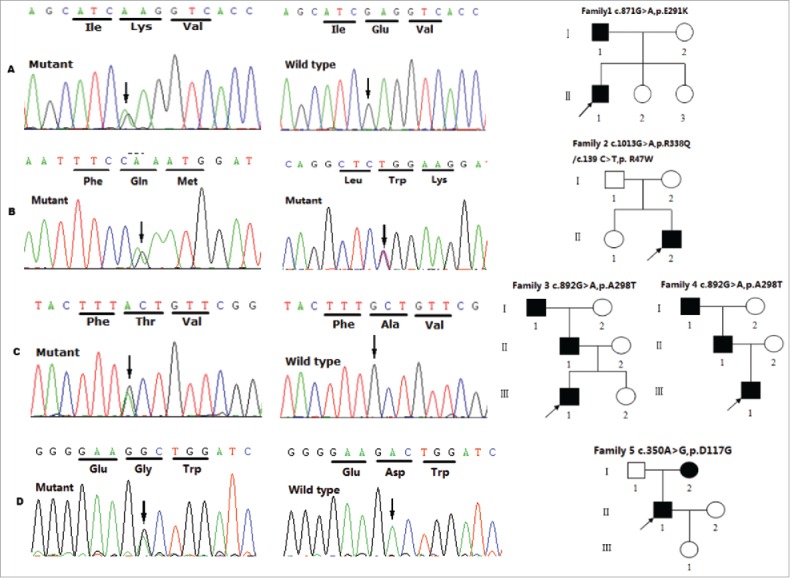
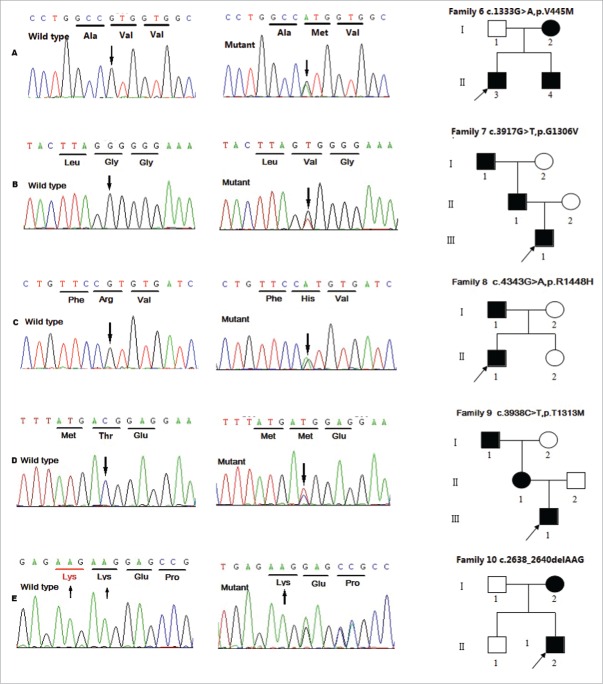
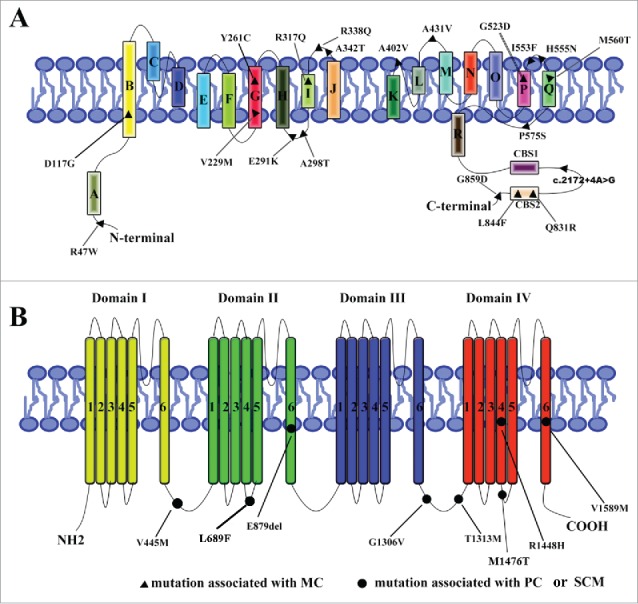
Myotonia congenita (MC), paramyotonia congenita (PC) and sodium channel myotonias(SCM) were belonged to Non-dystrophic myotonias, in which muscle relaxation is delayed after voluntary or evoked contraction. These diseases can not be simply distinguished only based on symptoms and signs but also on genetics: more than 100 mutations in the CLCN1 gene have been associated with MC, while at least 20 mutations in the SCN4A gene have been associated with PC and SCM. Most of these genetics studies have been conducted outside China, only several MC, PC, and SCM families accepted gene scan were reported in China. Therefore we analyzed genetic mutations in CLCN1 and SCN4A in 10 Chinese families clinically diagnosed with Non-dystrophic myotonias. Our result revealed 12 potential disease-causing mutations(3 mutations were novel) that were present in the probands and affected family members. We also reviewed all available literature on mutations linked to these 3 disease in Chinese populations. Our results may help identify genetic determinants as well as clarify genotype-phenotype relationships.
Keywords: CLCN1; SCN4A; myotonia congenita; paramyotonia congenita; sodium channel myotonias.
Figures
References
-
- Kubisch C, Schmidt-Rose T, Fontaine B, Bretag AH, Jentsch TJ. ClC-1 chloride channel mutations in myotonia congenita: variable penetrance of mutations shifting the voltage dependence. Hum Mol Genet 1998; 7:1753-60; PMID:9736777; http://dx.doi.org/ 10.1093/hmg/7.11.1753 - DOI - PubMed
-
- Lee SC, Kim HS, Park YE, Choi YC, Park KH, Kim DS. Clinical Diversity of SCN4A- Mutation- Associated Skeletal Muscle Sodium Channelopathy. J Clin Neurol 2009; 5:186-91; PMID:20076800; http://dx.doi.org/ 10.3988/jcn.2009.5.4.186 - DOI - PMC - PubMed
-
- Matthews E, Fialho D, Tan SV, Venance SL, Cannon SC, Sternberg D, Fontaine B, Amato AA, Barohn RJ, Griggs RC, et al.. The non-dystrophic myotonias: molecular pathogenesis, diagnosis and treatment. Brain 2010; 133:9-22; PMID:19917643; http://dx.doi.org/ 10.1093/brain/awp294 - DOI - PMC - PubMed
-
- Cannon SC, Brown RH Jr, Corey DP. Theoretical reconstruction of myotonia and paralysis caused by incomplete inactivation of sodium channels. Biophys J 1993; 65:270-88; PMID:8396455; http://dx.doi.org/ 10.1016/S0006-3495(93)81045-2 - DOI - PMC - PubMed
-
- Nurputra DK, Nakagawa T, Takeshima Y, Harahap IS, Morikawa S, Sakaeda T, Lai PS, Matsuo M, Takaoka Y, Nishio H. Paramyotonia congenita: from clinical diagnosis to in silico protein modeling analysis. Pediatr Int 2012; 54:602-12; PMID:22507243; http://dx.doi.org/ 10.1111/j.1442-200X.2012.03646.x - DOI - PubMed
MeSH terms
Substances
Supplementary concepts
LinkOut - more resources
Full Text Sources
Other Literature Sources