

Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions
- PMID: 27419873
- PMCID: PMC5172462
- DOI: 10.1016/j.cell.2016.06.044
Epigenomic Diversity in a Global Collection of Arabidopsis thaliana Accessions
Abstract
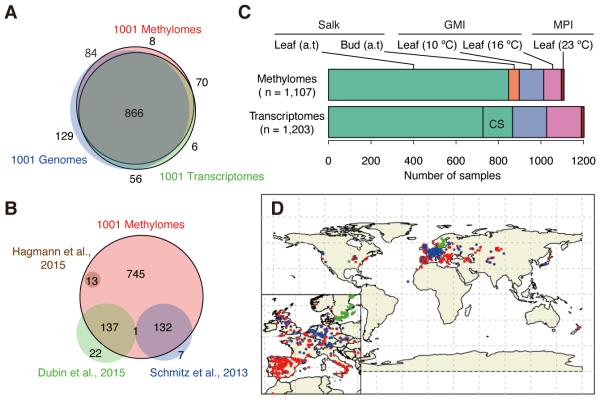
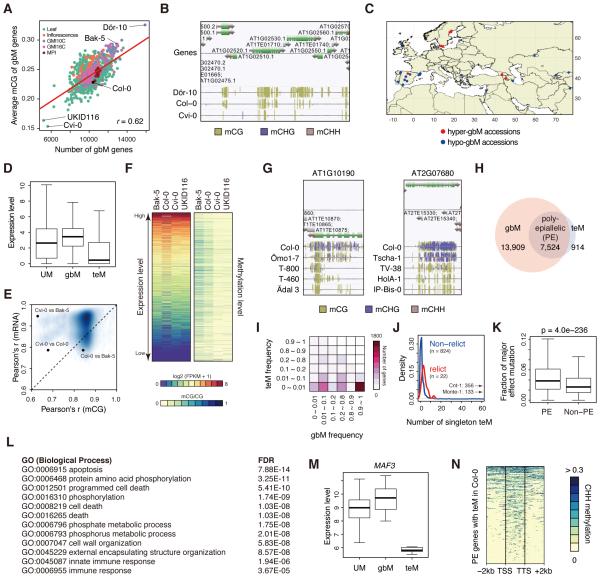
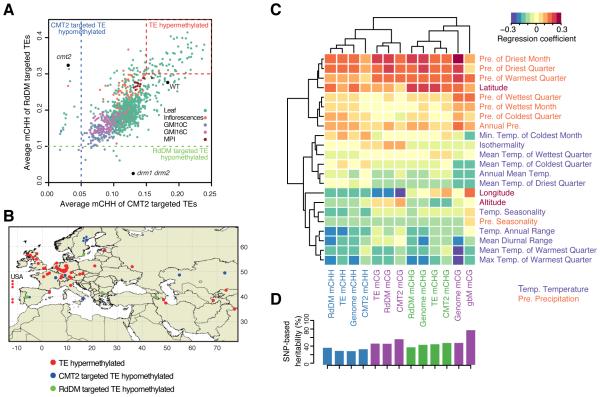
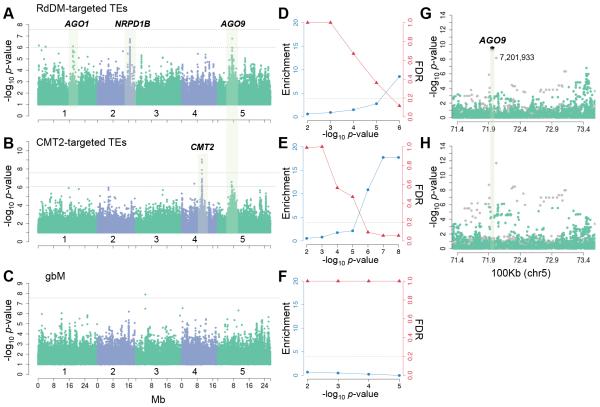
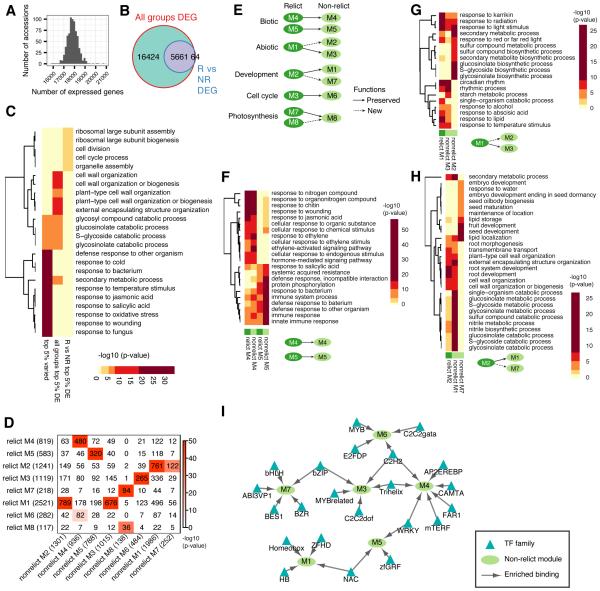
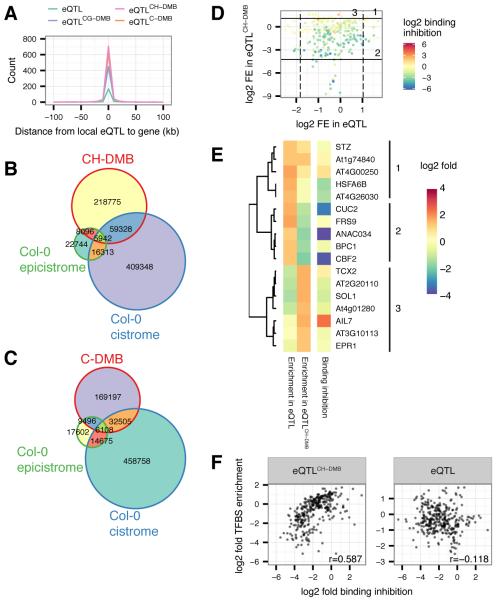
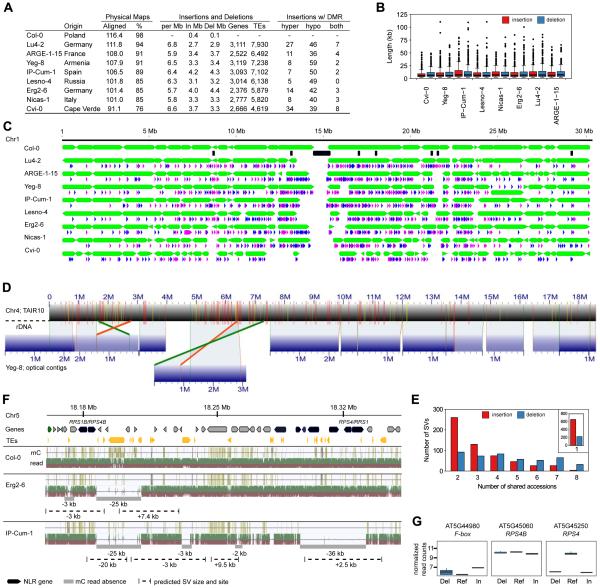
The epigenome orchestrates genome accessibility, functionality, and three-dimensional structure. Because epigenetic variation can impact transcription and thus phenotypes, it may contribute to adaptation. Here, we report 1,107 high-quality single-base resolution methylomes and 1,203 transcriptomes from the 1001 Genomes collection of Arabidopsis thaliana. Although the genetic basis of methylation variation is highly complex, geographic origin is a major predictor of genome-wide DNA methylation levels and of altered gene expression caused by epialleles. Comparison to cistrome and epicistrome datasets identifies associations between transcription factor binding sites, methylation, nucleotide variation, and co-expression modules. Physical maps for nine of the most diverse genomes reveal how transposons and other structural variants shape the epigenome, with dramatic effects on immunity genes. The 1001 Epigenomes Project provides a comprehensive resource for understanding how variation in DNA methylation contributes to molecular and non-molecular phenotypes in natural populations of the most studied model plant.
Copyright © 2016 Elsevier Inc. All rights reserved.
Figures
Comment in
-
Genomics: Geography matters for Arabidopsis.Nature. 2016 Sep 15;537(7620):314-315. doi: 10.1038/nature19466. Epub 2016 Sep 7. Nature. 2016. PMID: 27602515 No abstract available.
References
-
- Becker C, Hagmann J, Muller J, Koenig D, Stegle O, Borgwardt K, Weigel D. Spontaneous epigenetic variation in the Arabidopsis thaliana methylome. Nature. 2011;480:245–249. - PubMed
-
- Brodersen P, Sakvarelidze-Achard L, Bruun-Rasmussen M, Dunoyer P, Yamamoto YY, Sieburth L, Voinnet O. Widespread translational inhibition by plant miRNAs and siRNAs. Science. 2008;320:1185–1190. - PubMed
-
- Cao J, Schneeberger K, Ossowski S, Gunther T, Bender S, Fitz J, Koenig D, Lanz C, Stegle O, Lippert C, et al. Whole-genome sequencing of multiple Arabidopsis thaliana populations. Nat Genet. 2011;43:956–963. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
