

Consensus Genome-Wide Expression Quantitative Trait Loci and Their Relationship with Human Complex Trait Disease
- PMID: 27428252
- PMCID: PMC4960478
- DOI: 10.1089/omi.2016.0063
Consensus Genome-Wide Expression Quantitative Trait Loci and Their Relationship with Human Complex Trait Disease
Abstract
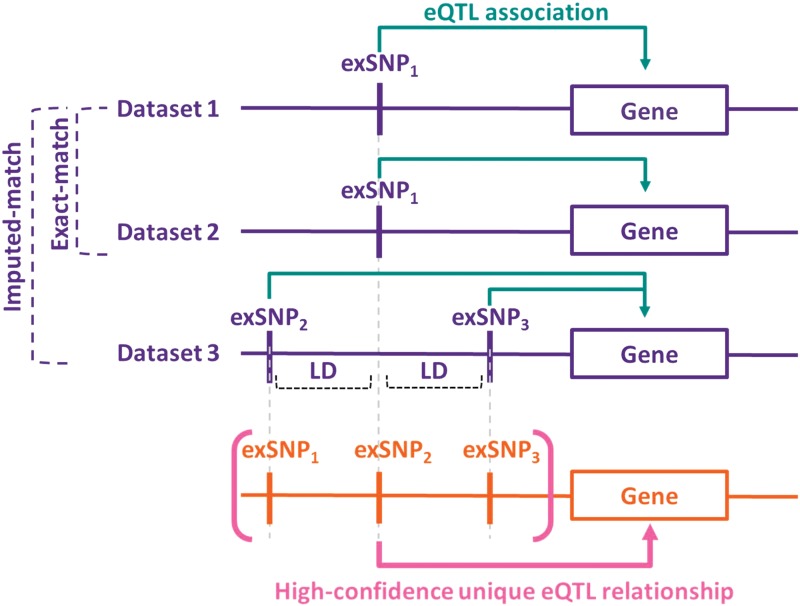
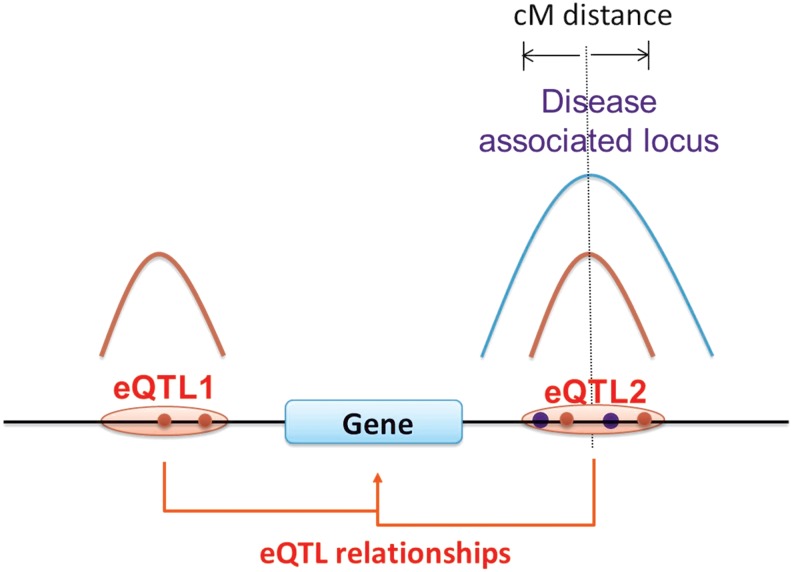
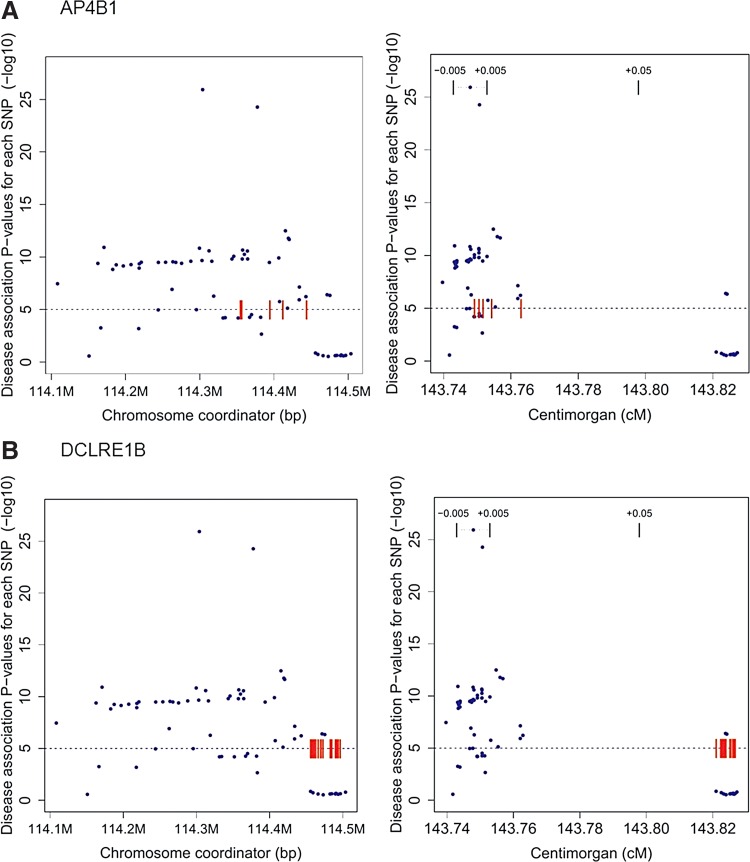
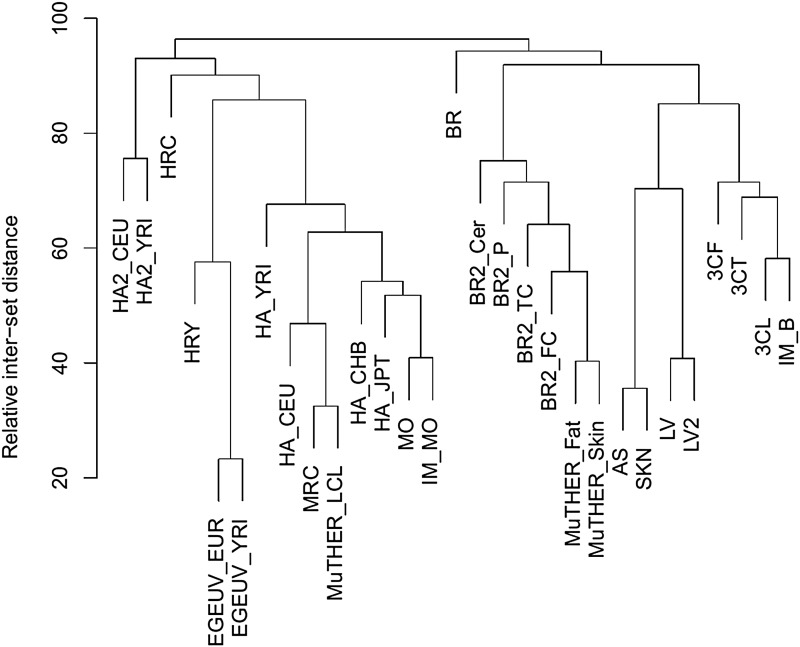
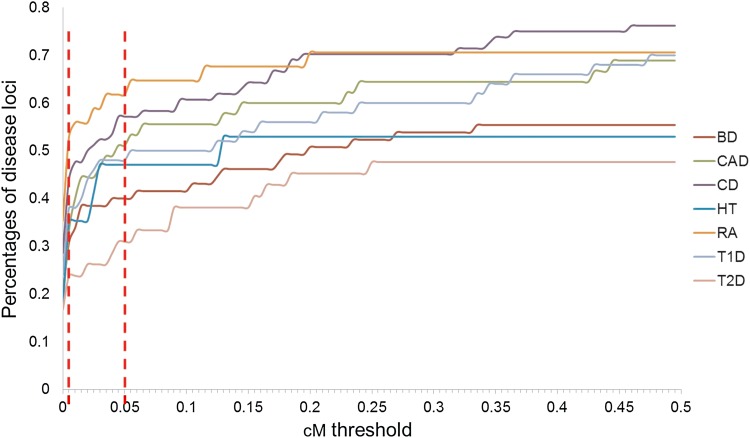
Most of the risk loci identified from genome-wide association (GWA) studies do not provide direct information on the biological basis of a disease or on the underlying mechanisms. Recent expression quantitative trait locus (eQTL) association studies have provided information on genetic factors associated with gene expression variation. These eQTLs might contribute to phenotype diversity and disease susceptibility, but interpretation is handicapped by low reproducibility of the expression results. To address this issue, we have generated a set of consensus eQTLs by integrating publicly available data for specific human populations and cell types. Overall, we find over 4000 genes that are involved in high-confidence eQTL relationships. To elucidate the role that eQTLs play in human common diseases, we matched the high-confidence eQTLs to a set of 335 disease risk loci identified from the Wellcome Trust Case Control Consortium GWA study and follow-up studies for 7 human complex trait diseases-bipolar disorder (BD), coronary artery disease (CAD), Crohn's disease (CD), hypertension (HT), rheumatoid arthritis (RA), type 1 diabetes (T1D), and type 2 diabetes (T2D). The results show that the data are consistent with ∼50% of these disease loci arising from an underlying expression change mechanism.
Figures
References
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Miscellaneous
