

ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks
- PMID: 27428653
- PMCID: PMC5003654
- DOI: 10.1038/nn.4345
ALS disrupts spinal motor neuron maturation and aging pathways within gene co-expression networks
Abstract
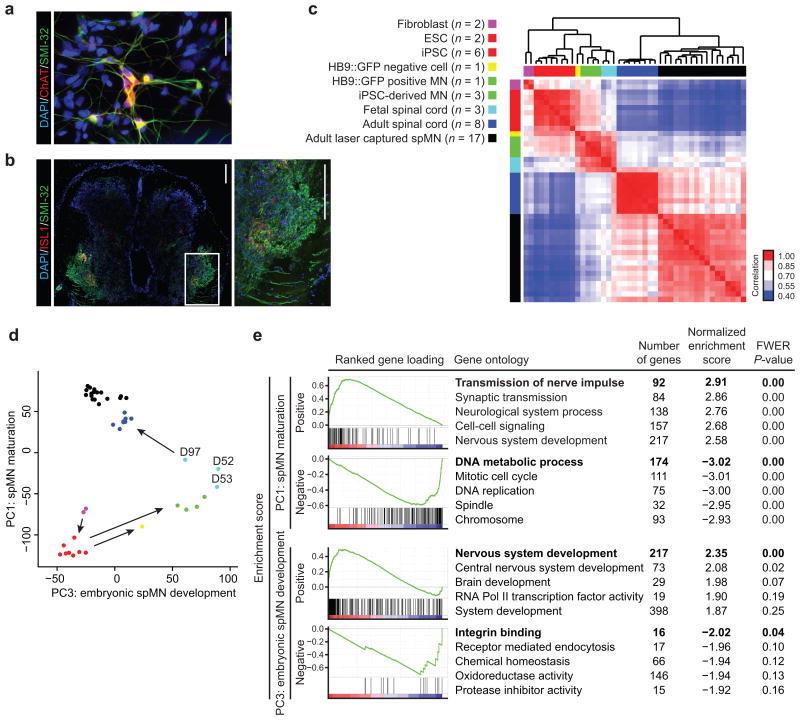
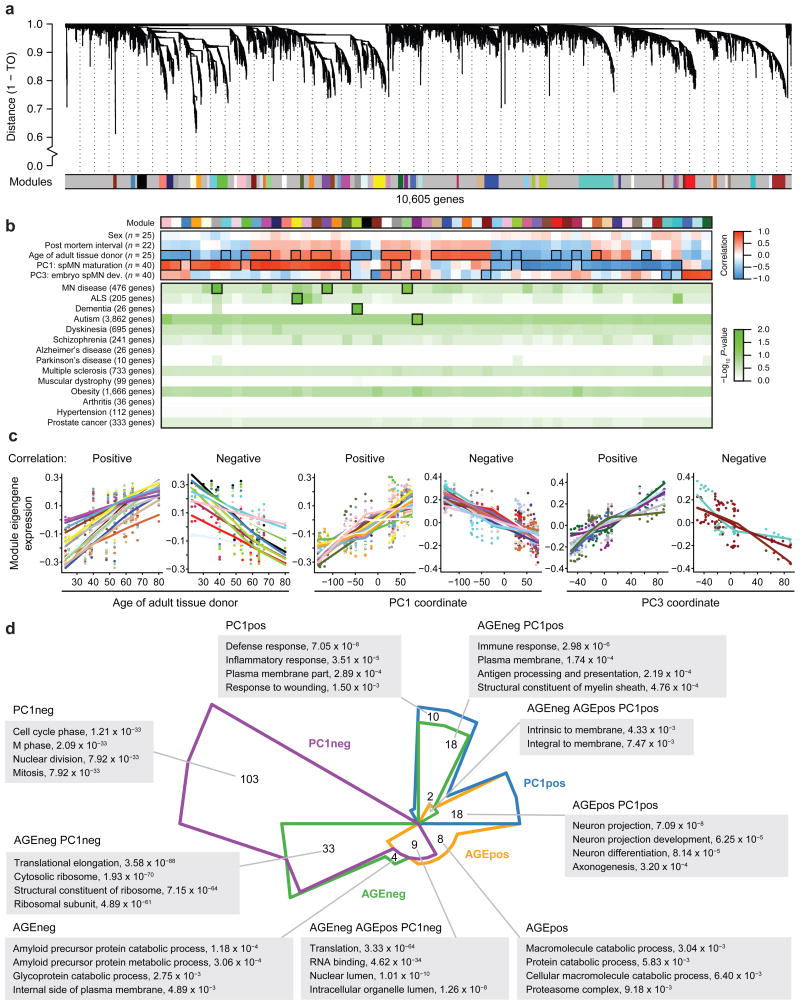
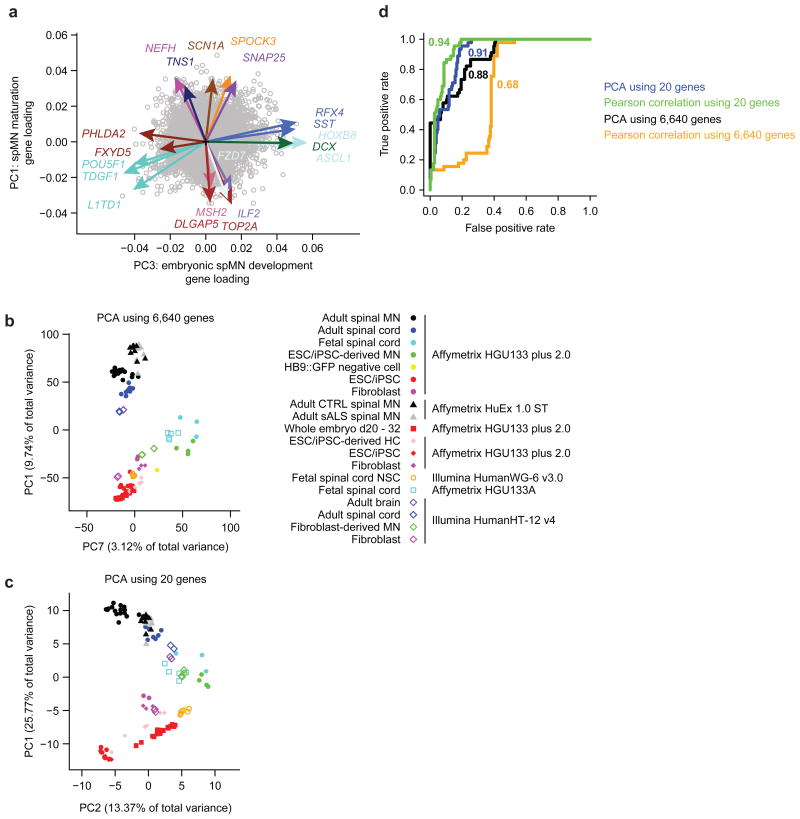
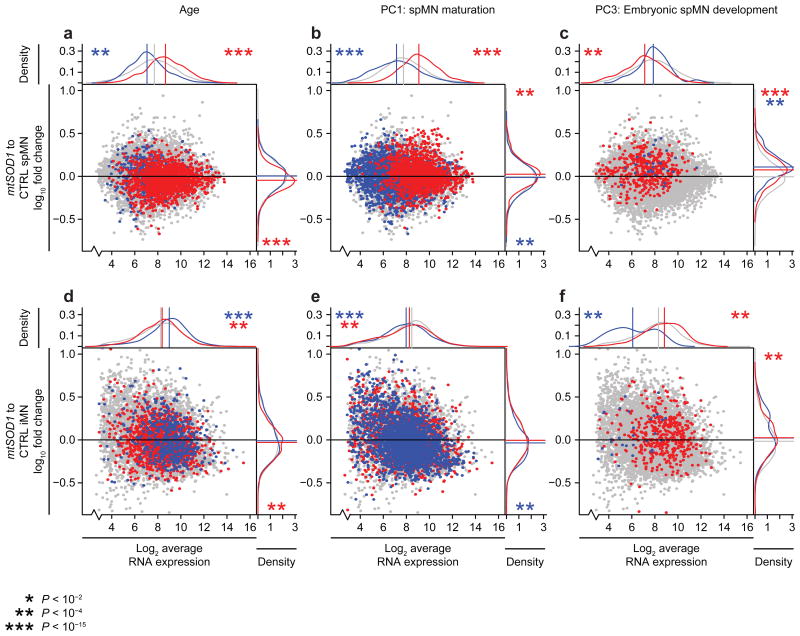
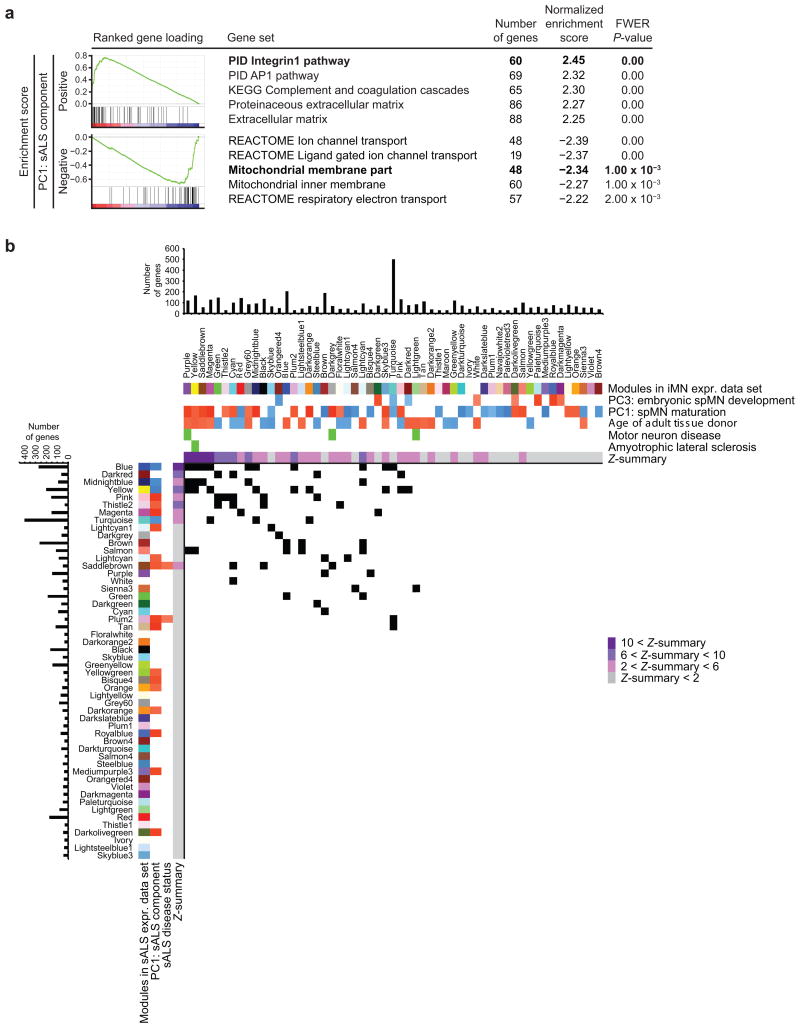
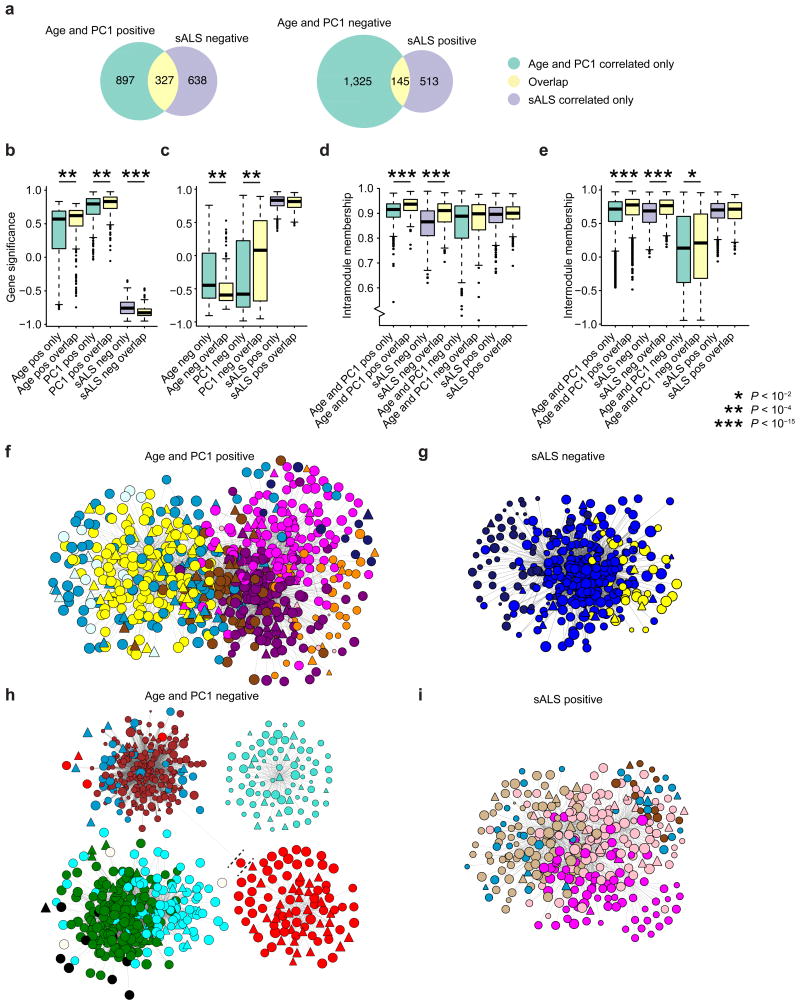
Modeling amyotrophic lateral sclerosis (ALS) with human induced pluripotent stem cells (iPSCs) aims to reenact embryogenesis, maturation and aging of spinal motor neurons (spMNs) in vitro. As the maturity of spMNs grown in vitro compared to spMNs in vivo remains largely unaddressed, it is unclear to what extent this in vitro system captures critical aspects of spMN development and molecular signatures associated with ALS. Here, we compared transcriptomes among iPSC-derived spMNs, fetal spinal tissues and adult spinal tissues. This approach produced a maturation scale revealing that iPSC-derived spMNs were more similar to fetal spinal tissue than to adult spMNs. Additionally, we resolved gene networks and pathways associated with spMN maturation and aging. These networks enriched for pathogenic familial ALS genetic variants and were disrupted in sporadic ALS spMNs. Altogether, our findings suggest that developing strategies to further mature and age iPSC-derived spMNs will provide more effective iPSC models of ALS pathology.
Figures
References
-
- Hardiman O, van den Berg LH, Kiernan MC. Clinical diagnosis and management of amyotrophic lateral sclerosis. Nat Rev Neurol. 2011;7(11):639–49. - PubMed
-
- Dimos JT, et al. Induced pluripotent stem cells generated from patients with ALS can be differentiated into motor neurons. Science. 2008;321(5893):1218–21. - PubMed
-
- Sterneckert JL, Reinhardt P, Scholer HR. Investigating human disease using stem cell models. Nat Rev Genet. 2014;15(9):625–39. - PubMed
-
- Jessell TM. Neuronal specification in the spinal cord: inductive signals and transcriptional codes. Nat Rev Genet. 2000;1(1):20–9. - PubMed
-
- Wichterle H, et al. Directed differentiation of embryonic stem cells into motor neurons. Cell. 2002;110(3):385–97. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
