

LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis
- PMID: 27430239
- PMCID: PMC5016083
- DOI: 10.1161/CIRCRESAHA.116.308955
LNK/SH2B3 Loss of Function Promotes Atherosclerosis and Thrombosis
Abstract
Rationale: Human genome-wide association studies have revealed novel genetic loci that are associated with coronary heart disease. One such locus resides in LNK/SH2B3, which in mice is expressed in hematopoietic cells and suppresses thrombopoietin signaling via its receptor myeloproliferative leukemia virus oncogene. However, the mechanisms underlying the association of LNK single-nucleotide polymorphisms with coronary heart disease are poorly understood.
Objective: To understand the functional effects of LNK single-nucleotide polymorphisms and explore the mechanisms whereby LNK loss of function impacts atherosclerosis and thrombosis.
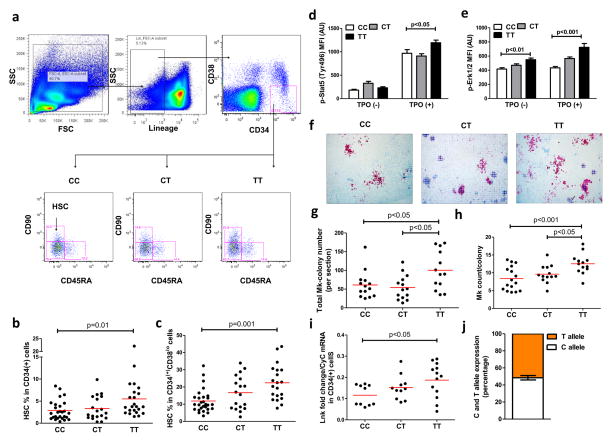
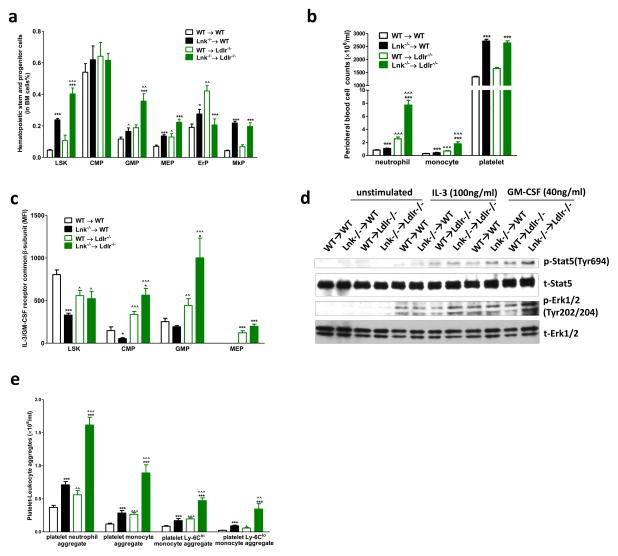
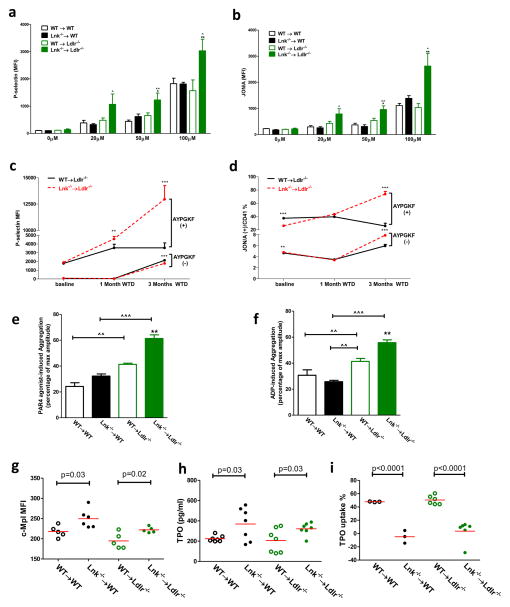
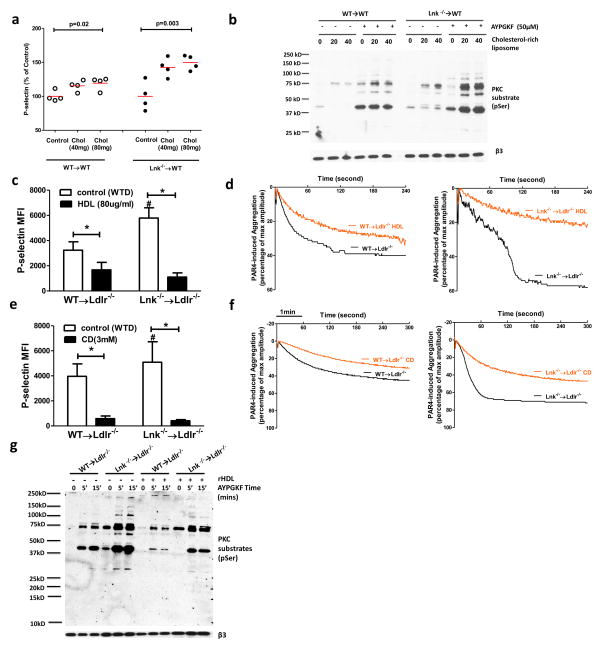
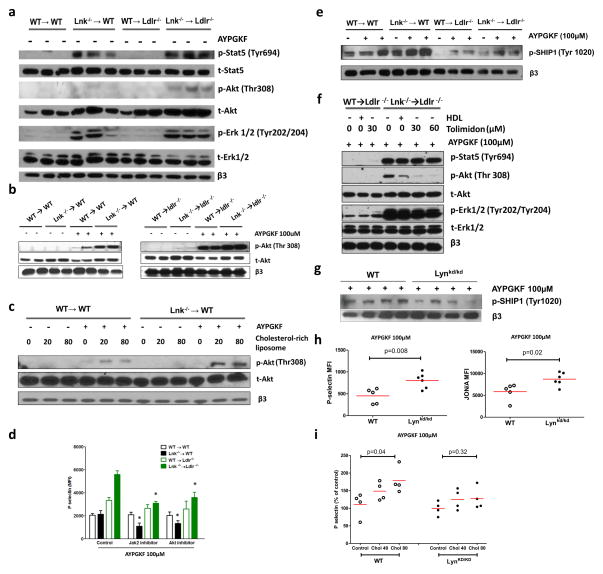
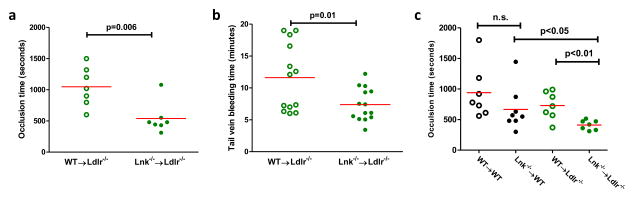
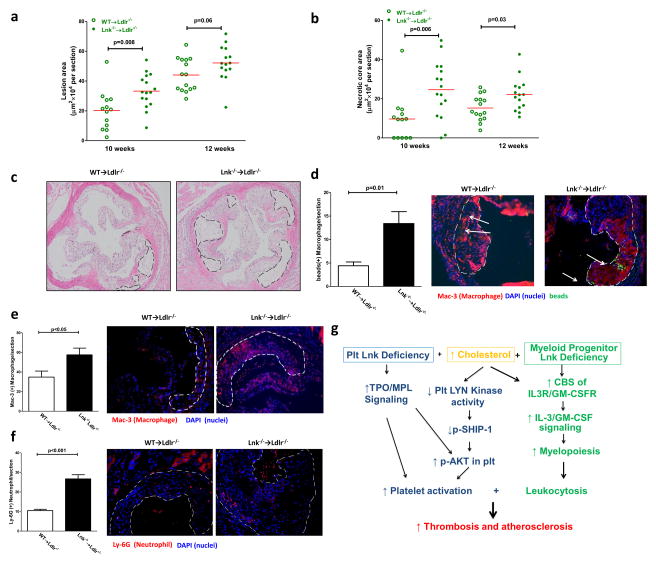
Methods and results: Using human cord blood, we show that the common TT risk genotype (R262W) of LNK is associated with expansion of hematopoietic stem cells and enhanced megakaryopoiesis, demonstrating reduced LNK function and increased myeloproliferative leukemia virus oncogene signaling. In mice, hematopoietic Lnk deficiency leads to accelerated arterial thrombosis and atherosclerosis, but only in the setting of hypercholesterolemia. Hypercholesterolemia acts synergistically with LNK deficiency to increase interleukin 3/granulocyte-macrophage colony-stimulating factor receptor signaling in bone marrow myeloid progenitors, whereas in platelets cholesterol loading combines with Lnk deficiency to increase activation. Platelet LNK deficiency increases myeloproliferative leukemia virus oncogene signaling and AKT activation, whereas cholesterol loading decreases SHIP-1 phosphorylation, acting convergently to increase AKT and platelet activation. Together with increased myelopoiesis, platelet activation promotes prothrombotic and proatherogenic platelet/leukocyte aggregate formation.
Conclusions: LNK (R262W) is a loss-of-function variant that promotes thrombopoietin/myeloproliferative leukemia virus oncogene signaling and platelet and leukocyte production. In mice, LNK deficiency is associated with both increased platelet production and activation. Hypercholesterolemia acts in platelets and hematopoietic progenitors to exacerbate thrombosis and atherosclerosis associated with LNK deficiency.
Keywords: atherosclerosis; cholesterol; hematopoiesis; hypercholesterolemia; thrombosis.
© 2016 American Heart Association, Inc.
Figures
Comment in
-
Bone Marrow Takes Center Stage in Cardiovascular Disease.Circ Res. 2016 Sep 2;119(6):701-3. doi: 10.1161/CIRCRESAHA.116.309584. Circ Res. 2016. PMID: 27587406 Free PMC article. No abstract available.
References
-
- Coller BS. Leukocytosis and ischemic vascular disease morbidity and mortality: is it time to intervene? Arterioscler Thromb Vasc Biol. 2005;25:658–70. - PubMed
-
- Lee CD, Folsom AR, Nieto FJ, Chambless LE, Shahar E, Wolfe DA. White blood cell count and incidence of coronary heart disease and ischemic stroke and mortality from cardiovascular disease in African-American and White men and women: atherosclerosis risk in communities study. American journal of epidemiology. 2001;154:758–64. - PubMed
-
- Wu Y, Wu H, Mueller C, Gibson CM, Murphy S, Shi Y, Xu G, Yang J. Baseline platelet count and clinical outcome in acute coronary syndrome. Circ J. 2012;76:704–11. - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
