

Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents
- PMID: 27432225
- PMCID: PMC5435293
- DOI: 10.1177/0271678X16659303
Requisite ischemia for spreading depolarization occurrence after subarachnoid hemorrhage in rodents
Abstract
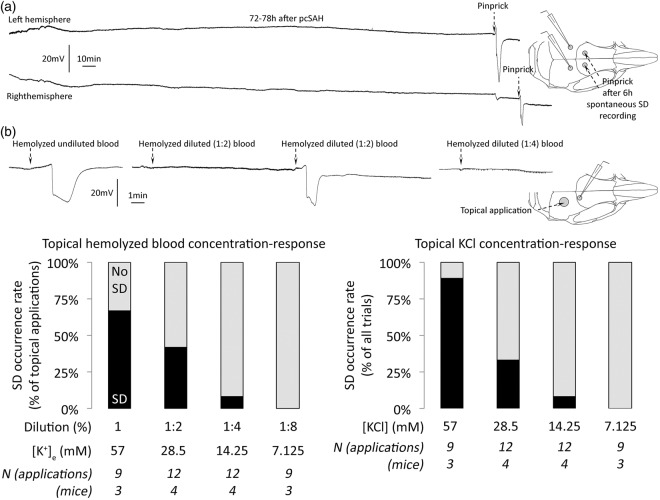
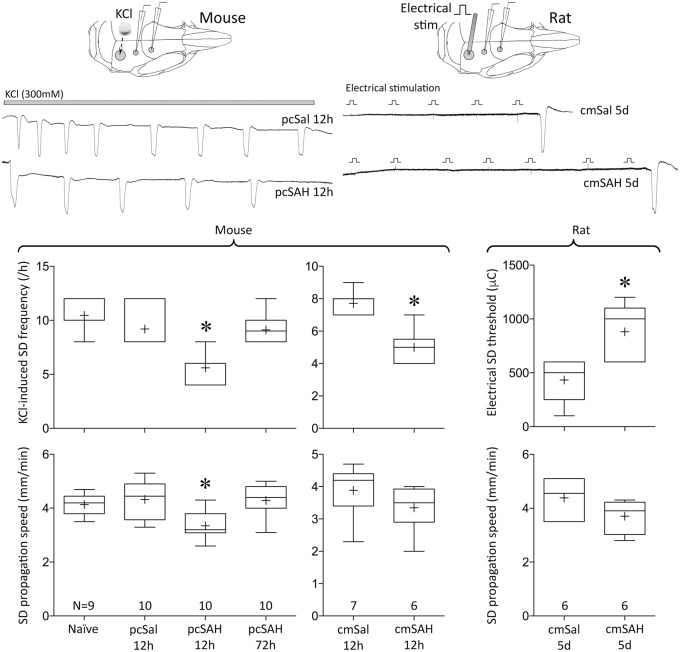
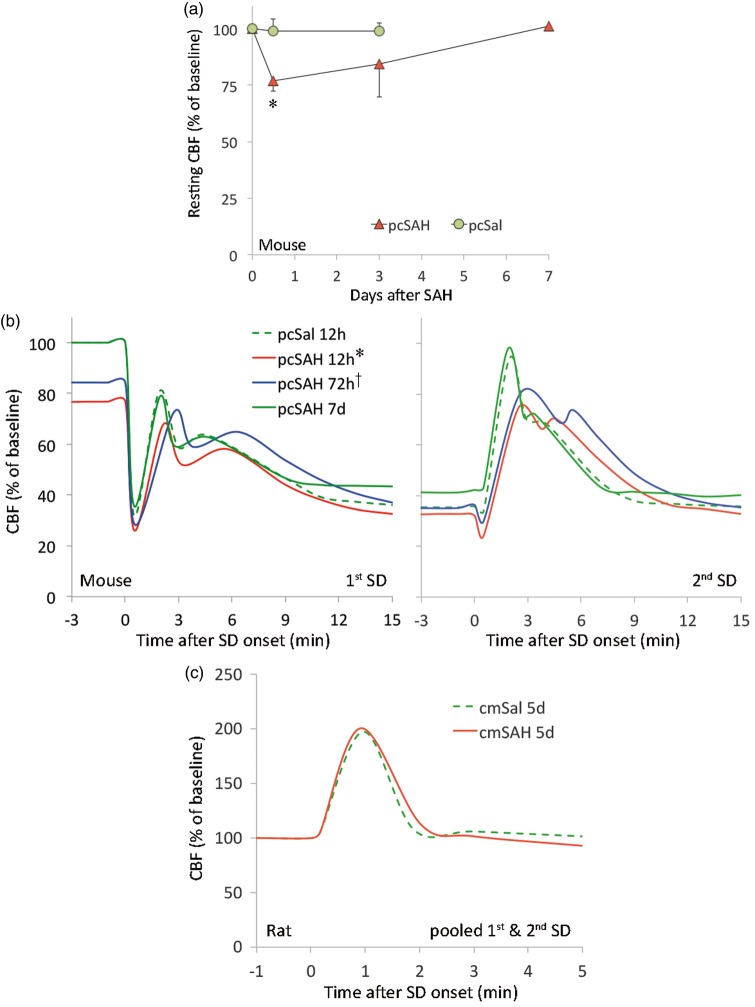
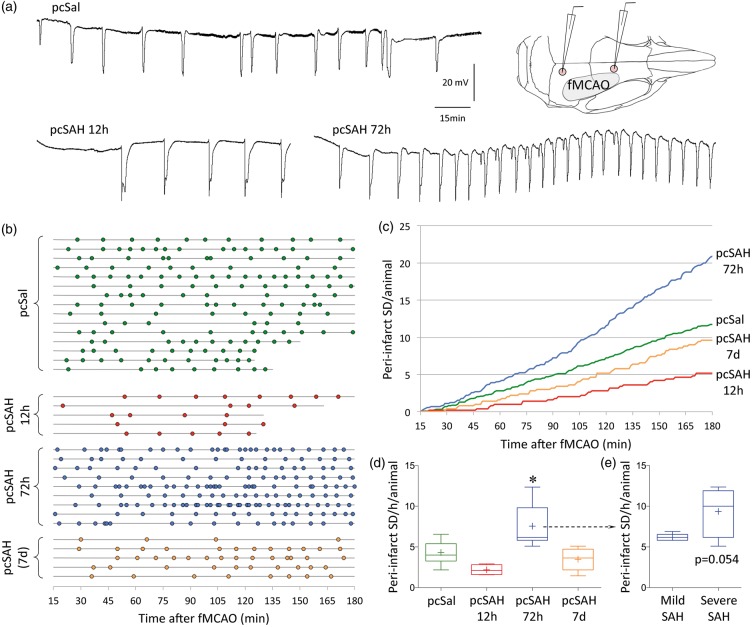
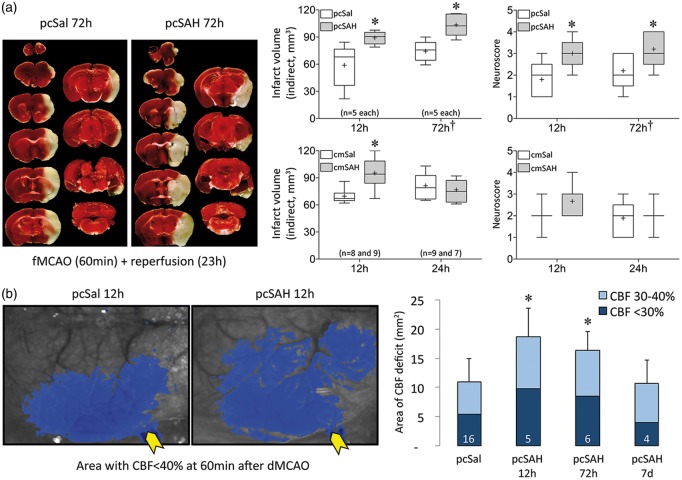
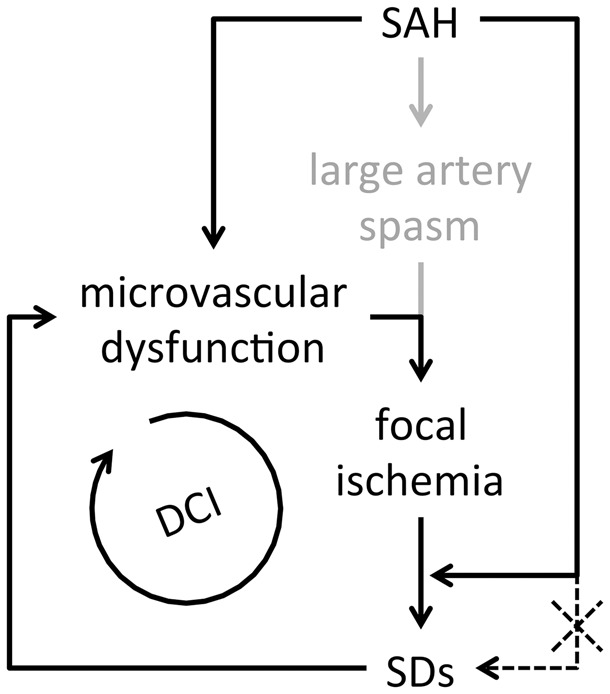
Spontaneous spreading depolarizations are frequent after various forms of human brain injury such as ischemic or hemorrhagic stroke and trauma, and worsen the outcome. We have recently shown that supply-demand mismatch transients trigger spreading depolarizations in ischemic stroke. Here, we examined the mechanisms triggering recurrent spreading depolarization events for many days after subarachnoid hemorrhage. Despite large volumes of subarachnoid hemorrhage induced by cisternal injection of fresh arterial blood in rodents, electrophysiological recordings did not detect a single spreading depolarization for up to 72 h after subarachnoid hemorrhage. Cortical susceptibility to spreading depolarization, measured by direct electrical stimulation or topical KCl application, was suppressed after subarachnoid hemorrhage. Focal cerebral ischemia experimentally induced after subarachnoid hemorrhage revealed a biphasic change in the propensity to develop peri-infarct spreading depolarizations. Frequency of peri-infarct spreading depolarizations decreased at 12 h, increased at 72 h and normalized at 7 days after subarachnoid hemorrhage compared with sham controls. However, ischemic tissue and neurological outcomes were significantly worse after subarachnoid hemorrhage even when peri-infarct spreading depolarization frequency was reduced. Laser speckle flowmetry implicated cerebrovascular hemodynamic mechanisms worsening the outcome. Altogether, our data suggest that cerebral ischemia is required for spreading depolarizations to be triggered after subarachnoid hemorrhage, which then creates a vicious cycle leading to the delayed cerebral ischemia syndrome.
Keywords: Subarachnoid hemorrhage; delayed cerebral ischemia; middle cerebral artery occlusion; peri-infarct depolarization; spreading depolarization.
Figures
Similar articles
-
Spreading depolarizations increase delayed brain injury in a rat model of subarachnoid hemorrhage.J Cereb Blood Flow Metab. 2016 Jul;36(7):1224-31. doi: 10.1177/0271678X15619189. Epub 2015 Nov 30. J Cereb Blood Flow Metab. 2016. PMID: 26661246 Free PMC article.
-
Advancing age and ischemia elevate the electric threshold to elicit spreading depolarization in the cerebral cortex of young adult rats.J Cereb Blood Flow Metab. 2017 May;37(5):1763-1775. doi: 10.1177/0271678X16643735. Epub 2016 Jan 1. J Cereb Blood Flow Metab. 2017. PMID: 27189902 Free PMC article.
-
Correlates of spreading depolarization in human scalp electroencephalography.Brain. 2012 Mar;135(Pt 3):853-68. doi: 10.1093/brain/aws010. Brain. 2012. PMID: 22366798 Free PMC article.
-
Cortical Spreading Depression and Ischemia in Neurocritical Patients.Neurosurg Clin N Am. 2018 Apr;29(2):223-229. doi: 10.1016/j.nec.2017.11.003. Neurosurg Clin N Am. 2018. PMID: 29502713 Review.
-
The role of spreading depression, spreading depolarization and spreading ischemia in neurological disease.Nat Med. 2011 Apr;17(4):439-47. doi: 10.1038/nm.2333. Epub 2011 Apr 7. Nat Med. 2011. PMID: 21475241 Review.
Cited by
-
Multimodal imaging of the role of hyperglycemia following experimental subarachnoid hemorrhage.J Cereb Blood Flow Metab. 2024 May;44(5):726-741. doi: 10.1177/0271678X231197946. Epub 2023 Sep 20. J Cereb Blood Flow Metab. 2024. PMID: 37728631 Free PMC article.
-
Is the Human Touch Always Therapeutic? Patient Stimulation and Spreading Depolarization after Acute Neurological Injuries.Transl Stroke Res. 2023 Apr;14(2):160-173. doi: 10.1007/s12975-022-01014-7. Epub 2022 Apr 2. Transl Stroke Res. 2023. PMID: 35364802 Free PMC article.
-
Rapid hematoma growth triggers spreading depolarizations in experimental intracortical hemorrhage.J Cereb Blood Flow Metab. 2021 Jun;41(6):1264-1276. doi: 10.1177/0271678X20951993. Epub 2020 Sep 16. J Cereb Blood Flow Metab. 2021. PMID: 32936730 Free PMC article.
-
Spreading Depolarizations and Subarachnoid Hemorrhage.Neurotherapeutics. 2020 Apr;17(2):497-510. doi: 10.1007/s13311-020-00850-5. Neurotherapeutics. 2020. PMID: 32323204 Free PMC article. Review.
-
Roles of glutamate in brain injuries after subarachnoid hemorrhage.Histol Histopathol. 2022 Nov;37(11):1041-1051. doi: 10.14670/HH-18-509. Epub 2022 Sep 6. Histol Histopathol. 2022. PMID: 36065974 Review.
References
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
