

Review
doi: 10.1084/jem.20160493.
Epub 2016 Jul 18.
Changes in insulin and insulin signaling in Alzheimer's disease: cause or consequence?
Affiliations
- PMID: 27432942
- PMCID: PMC4986537
- DOI: 10.1084/jem.20160493
Item in Clipboard
Review
Changes in insulin and insulin signaling in Alzheimer's disease: cause or consequence?
J Exp Med.
.
Abstract
Individuals with type 2 diabetes have an increased risk for developing Alzheimer's disease (AD), although the causal relationship remains poorly understood. Alterations in insulin signaling (IS) are reported in the AD brain. Moreover, oligomers/fibrils of amyloid-β (Aβ) can lead to neuronal insulin resistance and intranasal insulin is being explored as a potential therapy for AD. Conversely, elevated insulin levels (ins) are found in AD patients and high insulin has been reported to increase Aβ levels and tau phosphorylation, which could exacerbate AD pathology. Herein, we explore whether changes in ins and IS are a cause or consequence of AD.
© 2016 Stanley et al.
Figures
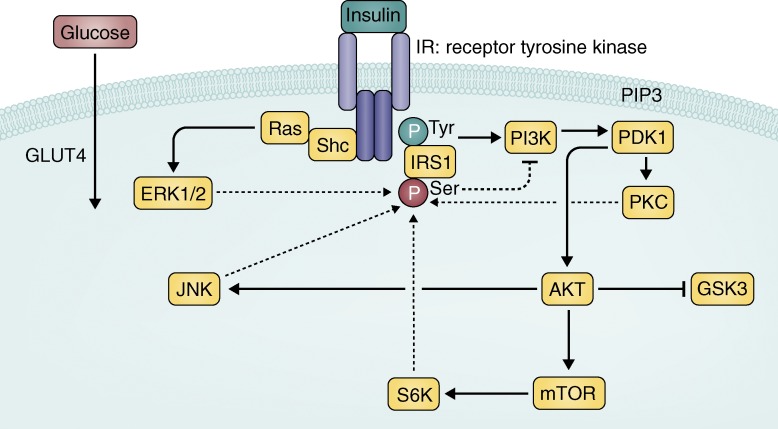
Canonical IR signaling cascade. Insulin binds to the insulin receptor
(IR), a receptor tyrosine kinase, which autophosphorylates and activates a cascade of
phosphorylation events. IRS1 is phosphorylated on a tyrosine residue to activate
further signaling, which ultimately leads to the translocation of glucose transporter
4 (GLUT4) to the membrane and uptake of glucose for energy in peripheral tissues.
Solid arrows represent activation upon insulin stimulation. Blocked arrows represent
inhibition. Glycogen synthase kinase 3 (GSK3) is serine phosphorylated and inhibited
in response to insulin stimulation. Dashed arrows represent downstream effectors that
have been found to phosphorylate IRS1 on a serine residue (p(Ser)-IRS1), which is
thought to lead to less activation of the signaling cascade through negative feedback
(dashed blocked arrow). p(Ser)-IRS1 is a marker of insulin resistance.
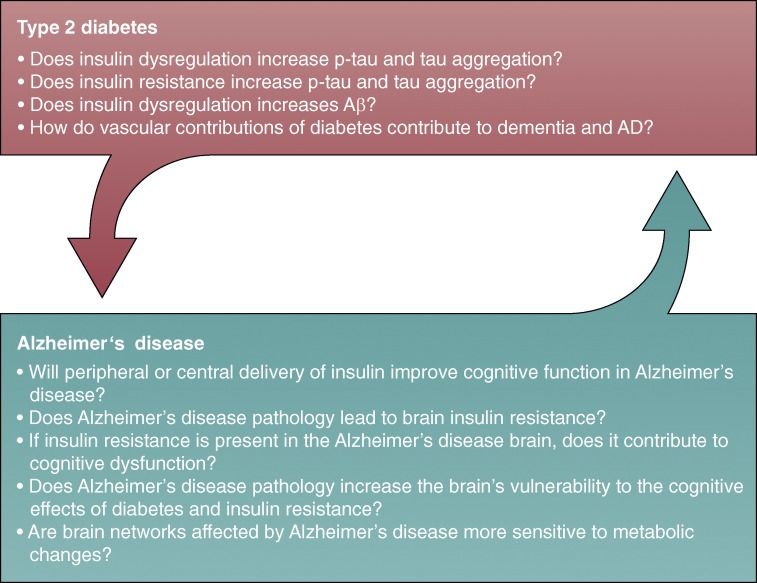
Connections between T2D and AD: cause or consequence? Big picture
questions that need to be addressed to determine if insulin-related changes represent
a cause or consequence of AD. In regards to the evidence that T2D increases the risk
of AD, answering the questions in the top arrow will determine how and why T2D is a
risk factor and the potentially causal role of insulin/IS. In regards to the idea
that AD progression may lead to a diabetic phenotype, answering the questions in the
bottom arrow will determine if and how AD pathology may affect insulin homeostasis
and the potential consequences of these changes on cognition.
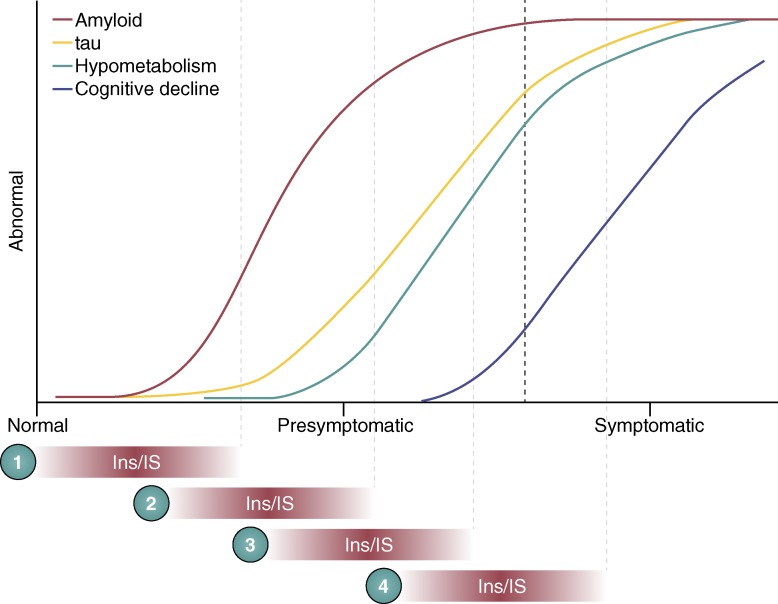
Where and when do changes in ins and IS affect AD? Colored lines
represent our current understanding of the pathological timeline in AD. It is
currently unclear when and where changes in ins/IS occur in this timeline. If
changes in ins/IS happen early, (1) they could initiate or potentiate amyloid
accumulation to casually influence AD. If ins/IS changes appear around the time
of symptoms (4), this could be a consequence of years of pathological changes
and may be directly related to cognitive decline. If changes in ins/IS occur in
the presymptomatic period (2 and 3), they could be interacting with Aβ,
tau, or metabolism to contribute to disease progression. Conversely,
presymptomatic changes could be a result of tau or Aβ accumulation or
metabolic perturbation. Additionally, it is possible that changes in ins/IS
could simply push the symptomatic period to the left (earlier) without directly
interacting with these pathologies.
References
-
- Baker L.D., Cross D.J., Minoshima S., Belongia D., Watson G.S., and Craft S.. 2011. Insulin resistance and Alzheimer-like reductions in regional cerebral glucose metabolism for cognitively normal adults with prediabetes or early type 2 diabetes. Arch. Neurol. 68:51–57. 10.1001/archneurol.2010.225 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
