

HIF2α-arginase axis is essential for the development of pulmonary hypertension
- PMID: 27432976
- PMCID: PMC4978263
- DOI: 10.1073/pnas.1602978113
HIF2α-arginase axis is essential for the development of pulmonary hypertension
Abstract
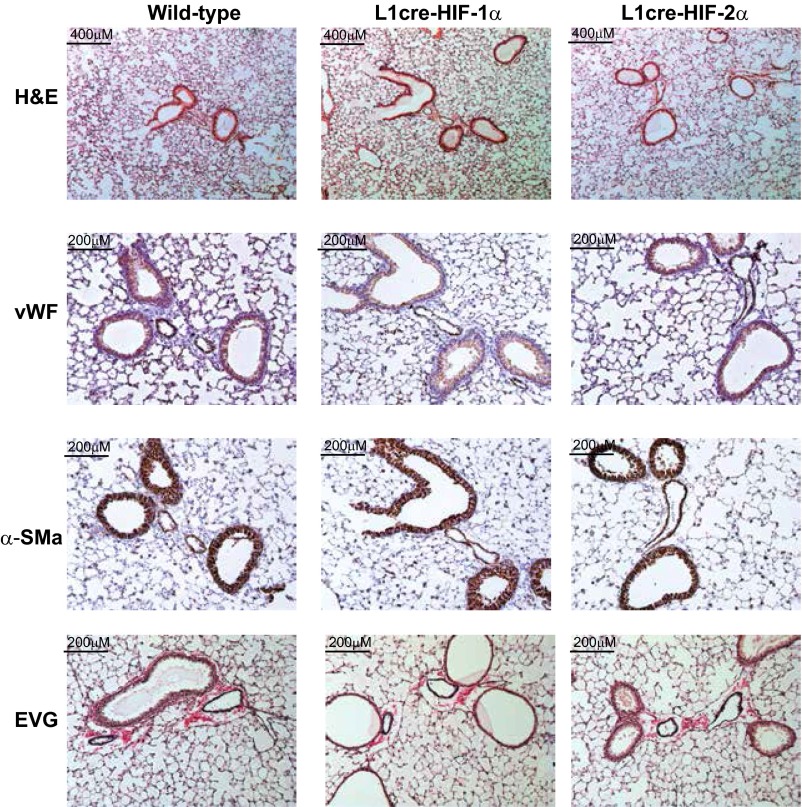
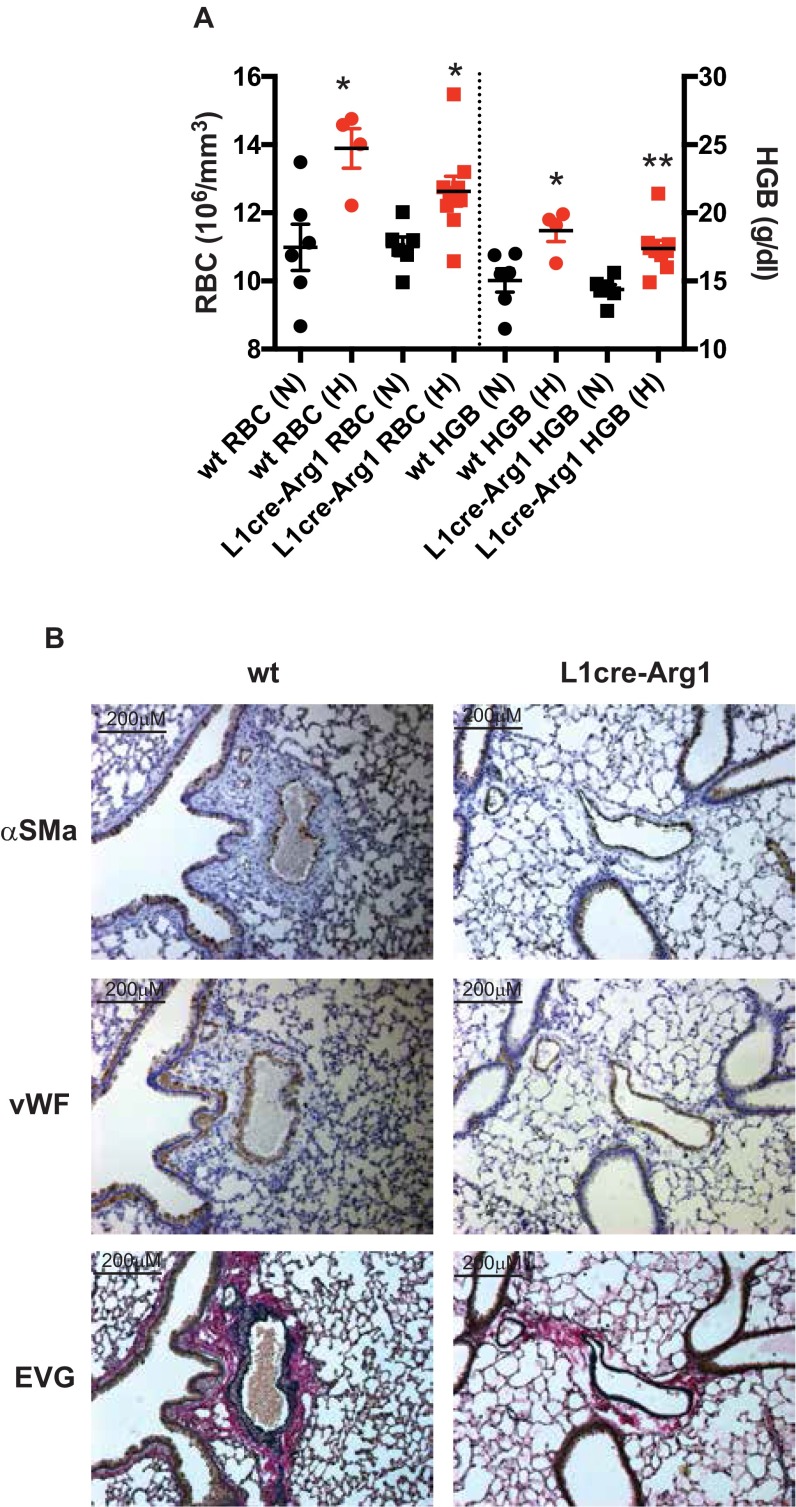
Hypoxic pulmonary vasoconstriction is correlated with pulmonary vascular remodeling. The hypoxia-inducible transcription factors (HIFs) HIF-1α and HIF-2α are known to contribute to the process of hypoxic pulmonary vascular remodeling; however, the specific role of pulmonary endothelial HIF expression in this process, and in the physiological process of vasoconstriction in response to hypoxia, remains unclear. Here we show that pulmonary endothelial HIF-2α is a critical regulator of hypoxia-induced pulmonary arterial hypertension. The rise in right ventricular systolic pressure (RVSP) normally observed following chronic hypoxic exposure was absent in mice with pulmonary endothelial HIF-2α deletion. The RVSP of mice lacking HIF-2α in pulmonary endothelium after exposure to hypoxia was not significantly different from normoxic WT mice and much lower than the RVSP values seen in WT littermate controls and mice with pulmonary endothelial deletion of HIF-1α exposed to hypoxia. Endothelial HIF-2α deletion also protected mice from hypoxia remodeling. Pulmonary endothelial deletion of arginase-1, a downstream target of HIF-2α, likewise attenuated many of the pathophysiological symptoms associated with hypoxic pulmonary hypertension. We propose a mechanism whereby chronic hypoxia enhances HIF-2α stability, which causes increased arginase expression and dysregulates normal vascular NO homeostasis. These data offer new insight into the role of pulmonary endothelial HIF-2α in regulating the pulmonary vascular response to hypoxia.
Keywords: HIF; hypertension; hypoxia; pulmonary.
Conflict of interest statement
The authors declare no conflict of interest.
Figures











Similar articles
-
Differential regulation of pulmonary vascular cell growth by hypoxia-inducible transcription factor-1α and hypoxia-inducible transcription factor-2α.Am J Respir Cell Mol Biol. 2013 Jul;49(1):78-85. doi: 10.1165/rcmb.2012-0107OC. Am J Respir Cell Mol Biol. 2013. PMID: 23492195 Free PMC article.
-
Endothelial HIF-2α contributes to severe pulmonary hypertension due to endothelial-to-mesenchymal transition.Am J Physiol Lung Cell Mol Physiol. 2018 Feb 1;314(2):L256-L275. doi: 10.1152/ajplung.00096.2017. Epub 2017 Oct 26. Am J Physiol Lung Cell Mol Physiol. 2018. PMID: 29074488 Free PMC article.
-
Role of Hypoxia-Inducible Factors in Regulating Right Ventricular Function and Remodeling during Chronic Hypoxia-induced Pulmonary Hypertension.Am J Respir Cell Mol Biol. 2020 Nov;63(5):652-664. doi: 10.1165/rcmb.2020-0023OC. Am J Respir Cell Mol Biol. 2020. PMID: 32692928 Free PMC article.
-
Multiplicity of hypoxia-inducible transcription factors and their connection to the circadian clock in the zebrafish.Physiol Biochem Zool. 2015 Mar-Apr;88(2):146-57. doi: 10.1086/679751. Epub 2015 Jan 14. Physiol Biochem Zool. 2015. PMID: 25730270 Review.
-
The Distinct Role of HIF-1α and HIF-2α in Hypoxia and Angiogenesis.Cells. 2025 May 4;14(9):673. doi: 10.3390/cells14090673. Cells. 2025. PMID: 40358197 Free PMC article. Review.
Cited by
-
Hypoxia-inducible factor signaling in pulmonary hypertension.J Clin Invest. 2020 Nov 2;130(11):5638-5651. doi: 10.1172/JCI137558. J Clin Invest. 2020. PMID: 32881714 Free PMC article. Review.
-
Paracrine Effects of Renal Proximal Tubular Epithelial Cells on Podocyte Injury under Hypoxic Conditions Are Mediated by Arginase-II and TGF-β1.Int J Mol Sci. 2023 Feb 10;24(4):3587. doi: 10.3390/ijms24043587. Int J Mol Sci. 2023. PMID: 36835007 Free PMC article.
-
Targeting Arginine in COVID-19-Induced Immunopathology and Vasculopathy.Metabolites. 2022 Mar 11;12(3):240. doi: 10.3390/metabo12030240. Metabolites. 2022. PMID: 35323682 Free PMC article. Review.
-
Metabolic reprogramming, oxidative stress, and pulmonary hypertension.Redox Biol. 2023 Aug;64:102797. doi: 10.1016/j.redox.2023.102797. Epub 2023 Jun 24. Redox Biol. 2023. PMID: 37392518 Free PMC article. Review.
-
Cardiovascular adaptation to hypoxia and the role of peripheral resistance.Elife. 2017 Oct 19;6:e28755. doi: 10.7554/eLife.28755. Elife. 2017. PMID: 29049022 Free PMC article.
References
-
- von Euler US, Liljestrand G. Observations on the pulmonary arterial blood pressure in the cat. Acta Physiol Scand. 1946;12(4):301–320.
-
- Naeije R. Pulmonary hypertension and right heart failure in chronic obstructive pulmonary disease. Proc Am Thorac Soc. 2005;2(1):20–22. - PubMed
-
- Lettieri CJ, Nathan SD, Barnett SD, Ahmad S, Shorr AF. Prevalence and outcomes of pulmonary arterial hypertension in advanced idiopathic pulmonary fibrosis. Chest. 2006;129(3):746–752. - PubMed
-
- Bärtsch P, Gibbs JS. Effect of altitude on the heart and the lungs. Circulation. 2007;116(19):2191–2202. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
