

Protein kinase C mechanisms that contribute to cardiac remodelling
- PMID: 27433023
- PMCID: PMC5024564
- DOI: 10.1042/CS20160036
Protein kinase C mechanisms that contribute to cardiac remodelling
Abstract
Protein phosphorylation is a highly-regulated and reversible process that is precisely controlled by the actions of protein kinases and protein phosphatases. Factors that tip the balance of protein phosphorylation lead to changes in a wide range of cellular responses, including cell proliferation, differentiation and survival. The protein kinase C (PKC) family of serine/threonine kinases sits at nodal points in many signal transduction pathways; PKC enzymes have been the focus of considerable attention since they contribute to both normal physiological responses as well as maladaptive pathological responses that drive a wide range of clinical disorders. This review provides a background on the mechanisms that regulate individual PKC isoenzymes followed by a discussion of recent insights into their role in the pathogenesis of diseases such as cancer. We then provide an overview on the role of individual PKC isoenzymes in the regulation of cardiac contractility and pathophysiological growth responses, with a focus on the PKC-dependent mechanisms that regulate pump function and/or contribute to the pathogenesis of heart failure.
Keywords: myocardial remodelling; post translational modification; protein kinase C.
© 2016 The Author(s). published by Portland Press Limited on behalf of the Biochemical Society.
Figures
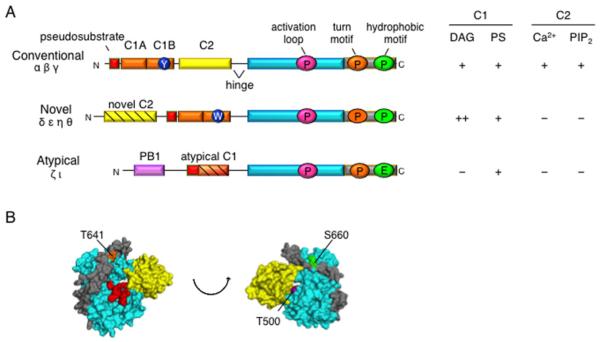
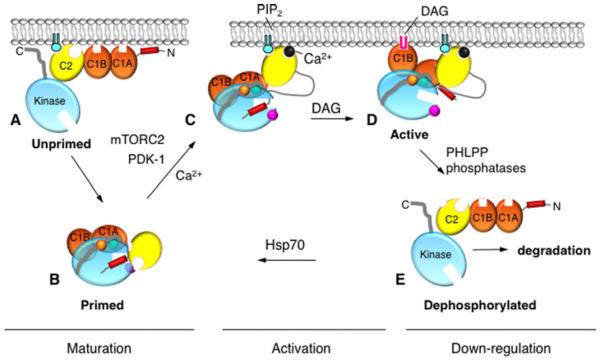
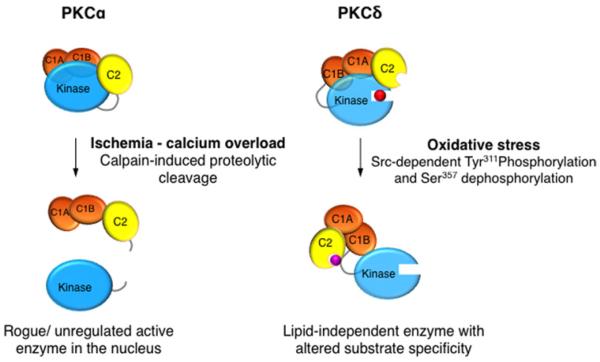
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources