

Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome
- PMID: 27434582
- PMCID: PMC4951053
- DOI: 10.1371/journal.pbio.1002511
Role of Mitochondrial Dynamics in Neuronal Development: Mechanism for Wolfram Syndrome
Abstract
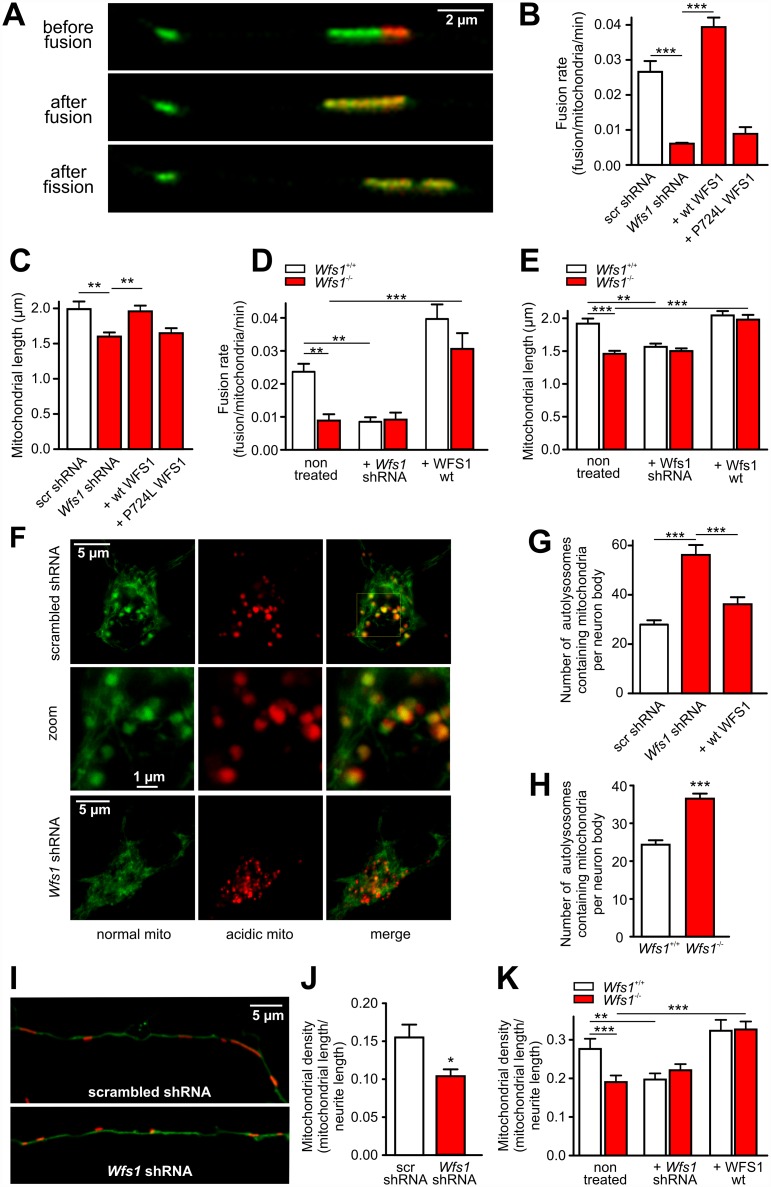
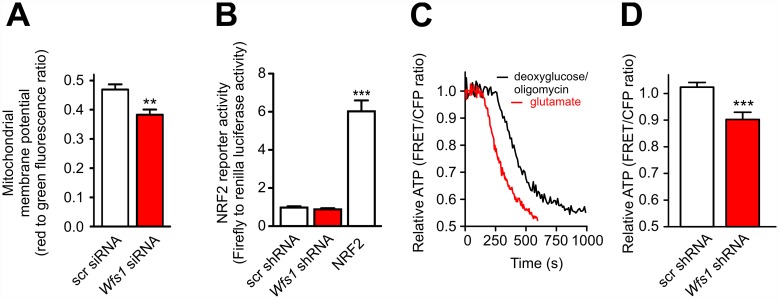
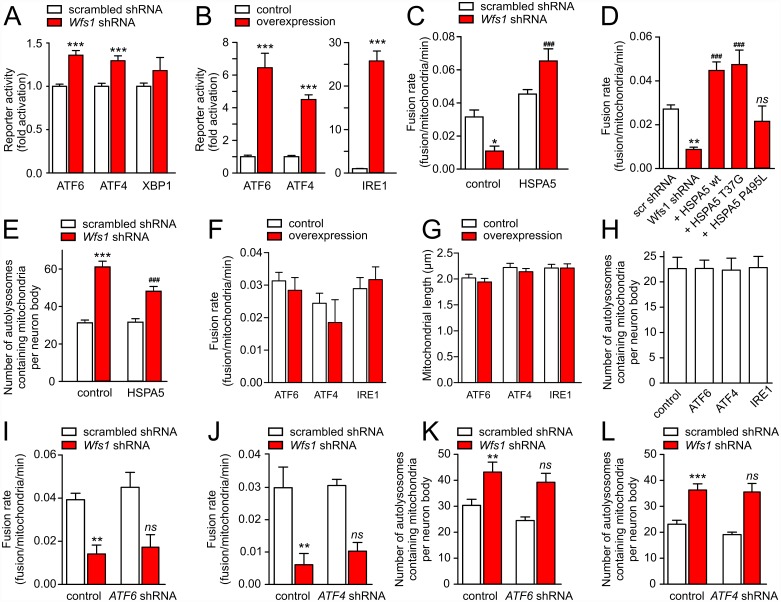
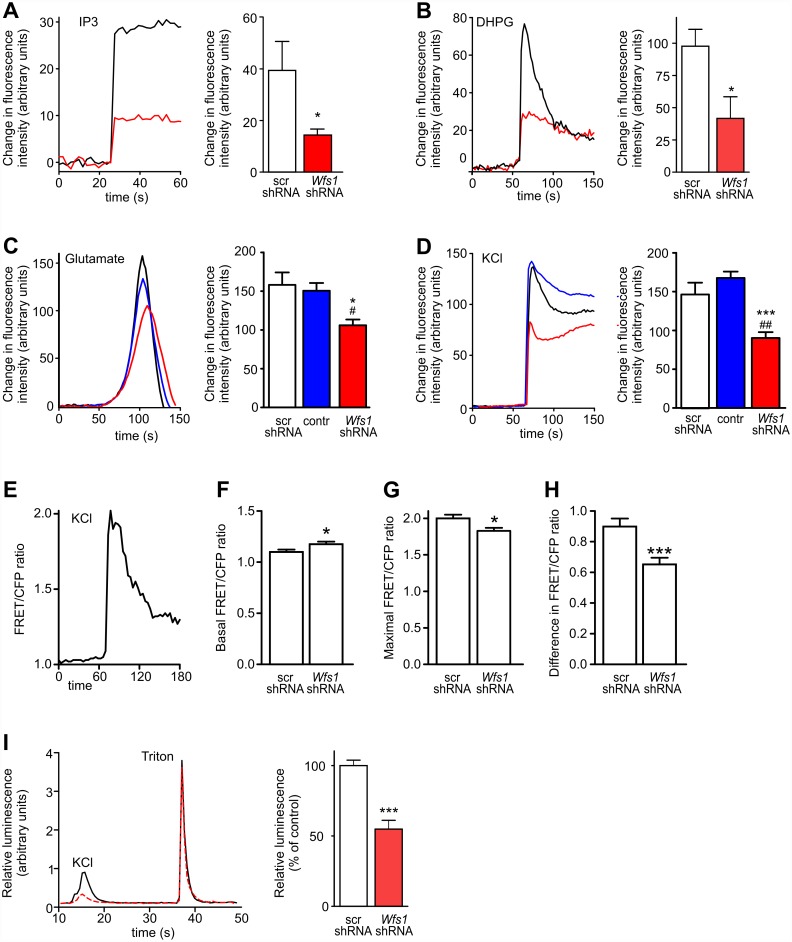
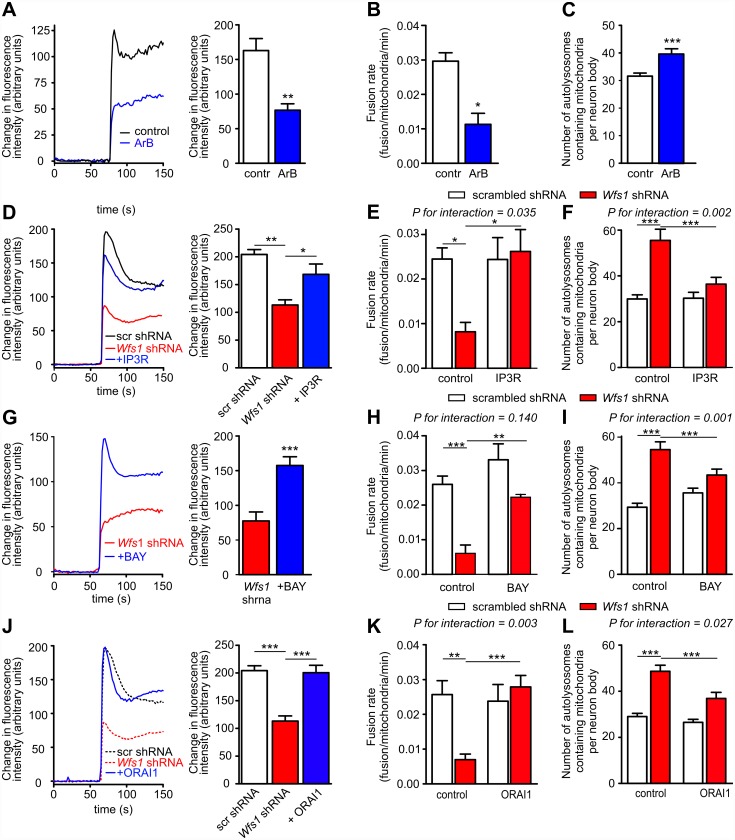
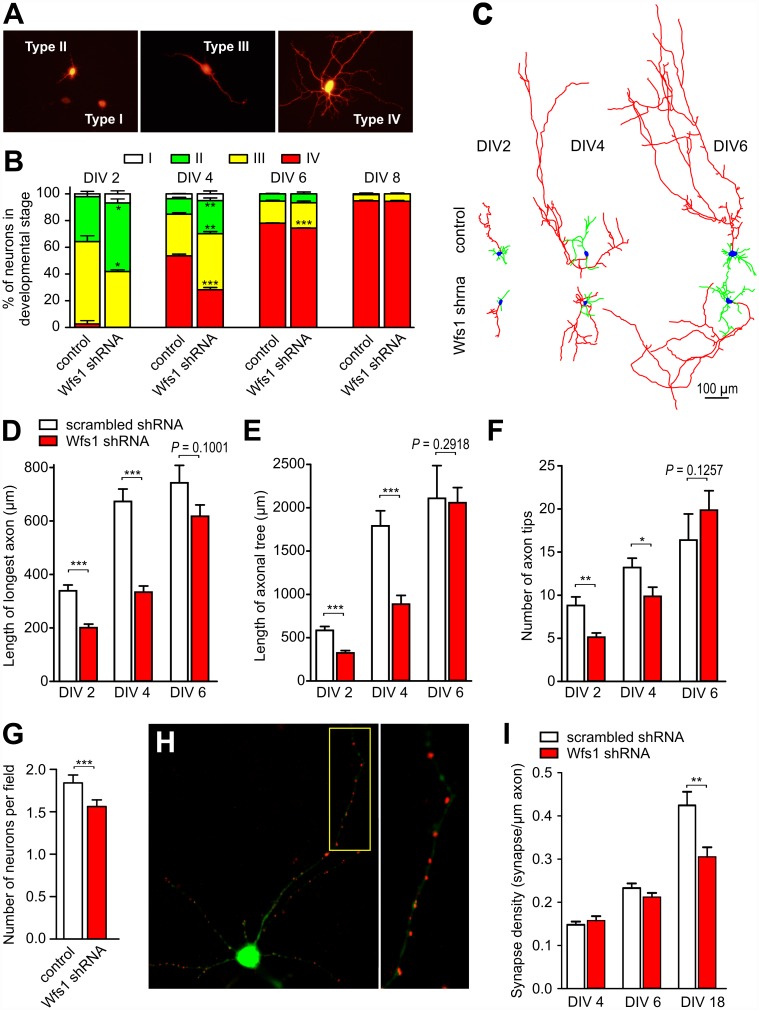
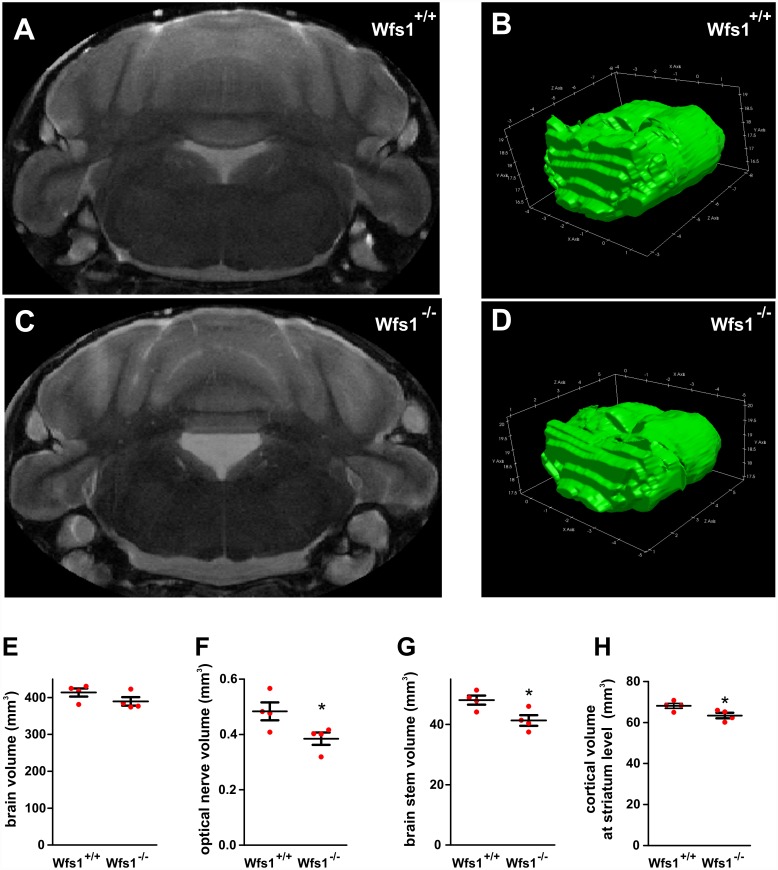
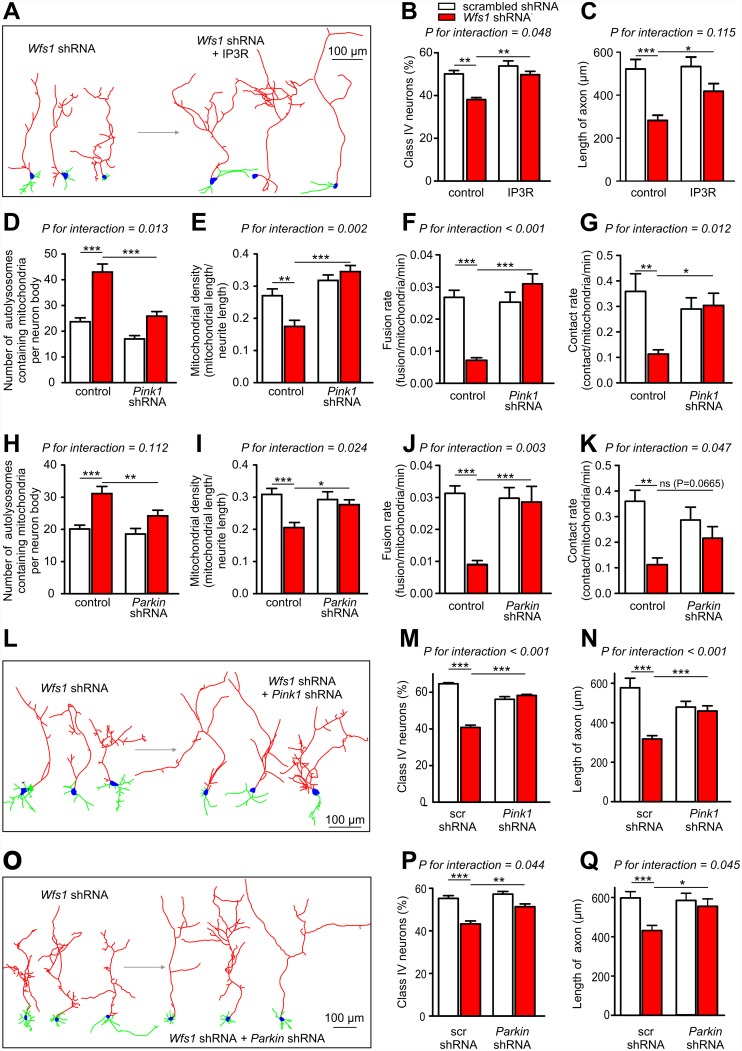
Deficiency of the protein Wolfram syndrome 1 (WFS1) is associated with multiple neurological and psychiatric abnormalities similar to those observed in pathologies showing alterations in mitochondrial dynamics. The aim of this study was to examine the hypothesis that WFS1 deficiency affects neuronal function via mitochondrial abnormalities. We show that down-regulation of WFS1 in neurons leads to dramatic changes in mitochondrial dynamics (inhibited mitochondrial fusion, altered mitochondrial trafficking, and augmented mitophagy), delaying neuronal development. WFS1 deficiency induces endoplasmic reticulum (ER) stress, leading to inositol 1,4,5-trisphosphate receptor (IP3R) dysfunction and disturbed cytosolic Ca2+ homeostasis, which, in turn, alters mitochondrial dynamics. Importantly, ER stress, impaired Ca2+ homeostasis, altered mitochondrial dynamics, and delayed neuronal development are causatively related events because interventions at all these levels improved the downstream processes. Our data shed light on the mechanisms of neuronal abnormalities in Wolfram syndrome and point out potential therapeutic targets. This work may have broader implications for understanding the role of mitochondrial dynamics in neuropsychiatric diseases.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Barrett TG, Bundey SE, Fielder AR, Good PA. Optic atrophy in Wolfram (DIDMOAD) syndrome. Eye. 1997;11:882–888. - PubMed
-
- Osman AA, Saito M, Makepeace C, Permutt MA, Schlesinger P, Mueckler MJ. Wolframin expression induces novel ion channel activity in endoplasmic reticulum membranes and increases intracellular calcium. J Biol Chem. 2003;278:52755–52762. - PubMed
-
- Takei D, Ishihara H, Yamaguchi S, Yamada T, Tamura A, Katagiri H, et al. WFS1 protein modulates the free Ca(2+) concentration in the endoplasmic reticulum. FEBS Lett. 2006;580:5635–5640. - PubMed
-
- Fonseca SG, Fukuma M, Lipson KL, Nguyen LX, Allen JR, Oka Y, et al. WFS1 is a novel component of the unfolded protein response and maintains homeostasis of the endoplasmic reticulum in pancreatic beta-cells. J Biol Chem. 2005;280:39609–39615. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
