

Structural Architecture of Prothrombin in Solution Revealed by Single Molecule Spectroscopy
- PMID: 27435675
- PMCID: PMC5000060
- DOI: 10.1074/jbc.M116.738310
Structural Architecture of Prothrombin in Solution Revealed by Single Molecule Spectroscopy
Abstract
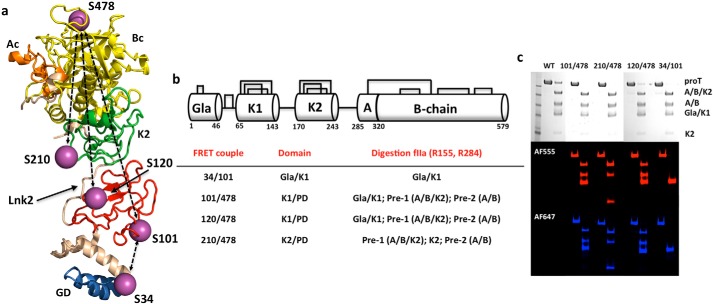
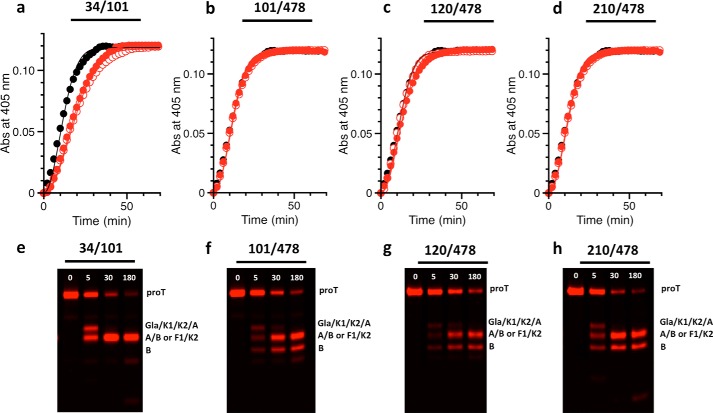
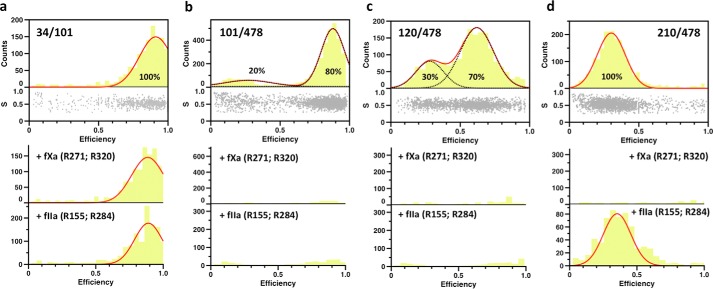
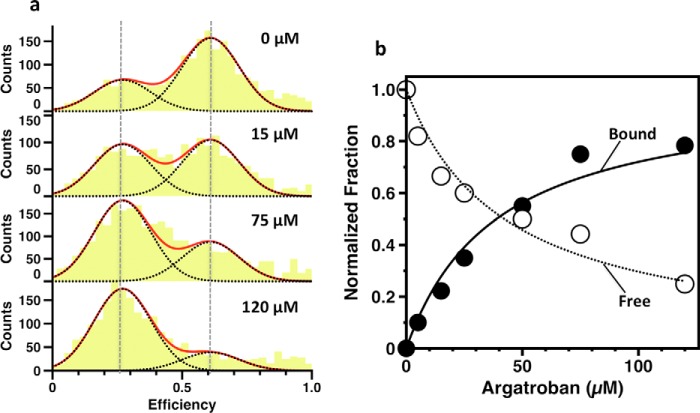
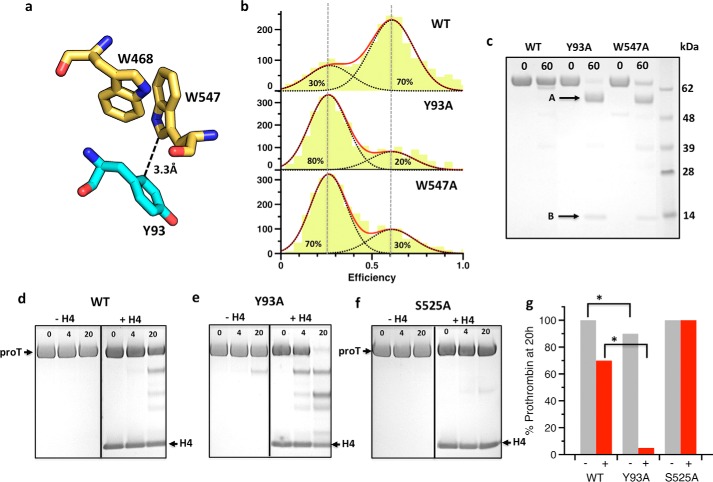
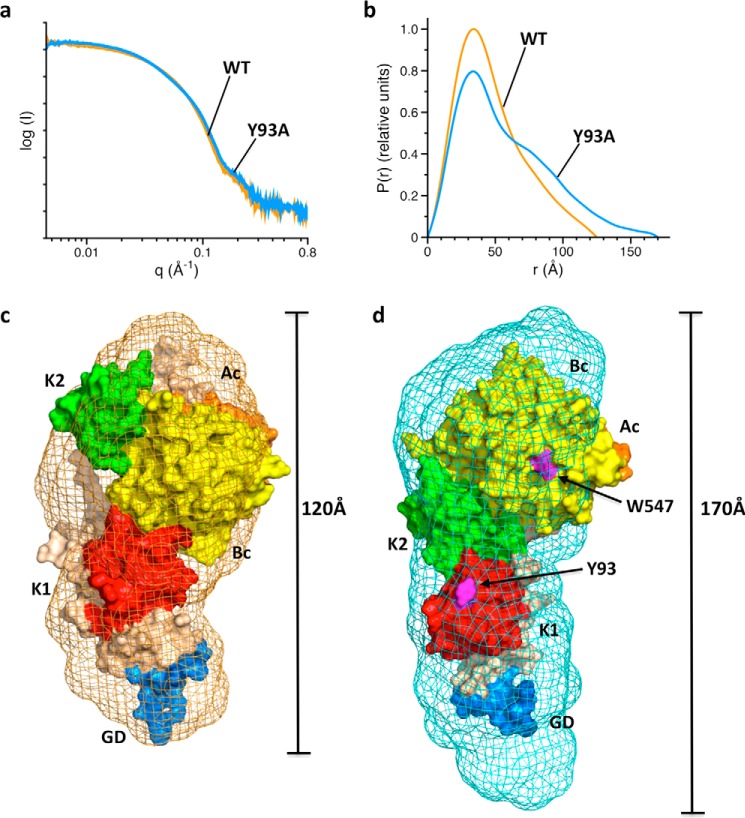
The coagulation factor prothrombin has a complex spatial organization of its modular assembly that comprises the N-terminal Gla domain, kringle-1, kringle-2, and the C-terminal protease domain connected by three intervening linkers. Here we use single molecule Förster resonance energy transfer to access the conformational landscape of prothrombin in solution and uncover structural features of functional significance that extend recent x-ray crystallographic analysis. Prothrombin exists in equilibrium between two alternative conformations, open and closed. The closed conformation predominates (70%) and features an unanticipated intramolecular collapse of Tyr(93) in kringle-1 onto Trp(547) in the protease domain that obliterates access to the active site and protects the zymogen from autoproteolytic conversion to thrombin. The open conformation (30%) is more susceptible to chymotrypsin digestion and autoactivation, and features a shape consistent with recent x-ray crystal structures. Small angle x-ray scattering measurements of prothrombin wild type stabilized 70% in the closed conformation and of the mutant Y93A stabilized 80% in the open conformation directly document two envelopes that differ 50 Å in length. These findings reveal important new details on the conformational plasticity of prothrombin in solution and the drastic structural difference between its alternative conformations. Prothrombin uses the intramolecular collapse of kringle-1 onto the active site in the closed form to prevent autoactivation. The open-closed equilibrium also defines a new structural framework for the mechanism of activation of prothrombin by prothrombinase.
Keywords: enzyme kinetics; prothrombin; single-molecule biophysics; structure-function; thrombin.
© 2016 by The American Society for Biochemistry and Molecular Biology, Inc.
Figures
References
-
- Krem M. M., and Di Cera E. (2002) Evolution of enzyme cascades from embryonic development to blood coagulation. Trends Biochem. Sci. 27, 67–74 - PubMed
-
- Patthy L. (1985) Evolution of the proteases of blood coagulation and fibrinolysis by assembly from modules. Cell 41, 657–663 - PubMed
-
- Papagrigoriou E., McEwan P. A., Walsh P. N., and Emsley J. (2006) Crystal structure of the factor XI zymogen reveals a pathway for transactivation. Nat. Struct. Mol. Biol. 13, 557–558 - PubMed
-
- Banner D. W., D'Arcy A., Chène C., Winkler F. K., Guha A., Konigsberg W. H., Nemerson Y., and Kirchhofer D. (1996) The crystal structure of the complex of blood coagulation factor VIIa with soluble tissue factor. Nature 380, 41–46 - PubMed
Publication types
MeSH terms
Substances
Associated data
- Actions
- Actions
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
