

Excess of Deleterious Mutations around HLA Genes Reveals Evolutionary Cost of Balancing Selection
- PMID: 27436009
- PMCID: PMC5026253
- DOI: 10.1093/molbev/msw127
Excess of Deleterious Mutations around HLA Genes Reveals Evolutionary Cost of Balancing Selection
Abstract
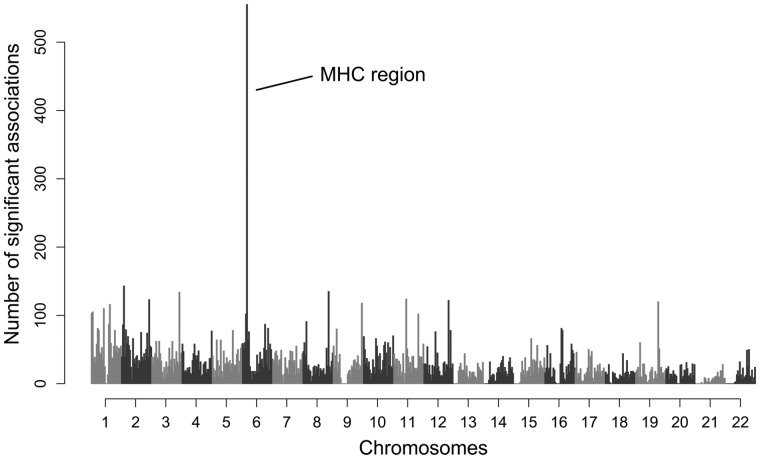
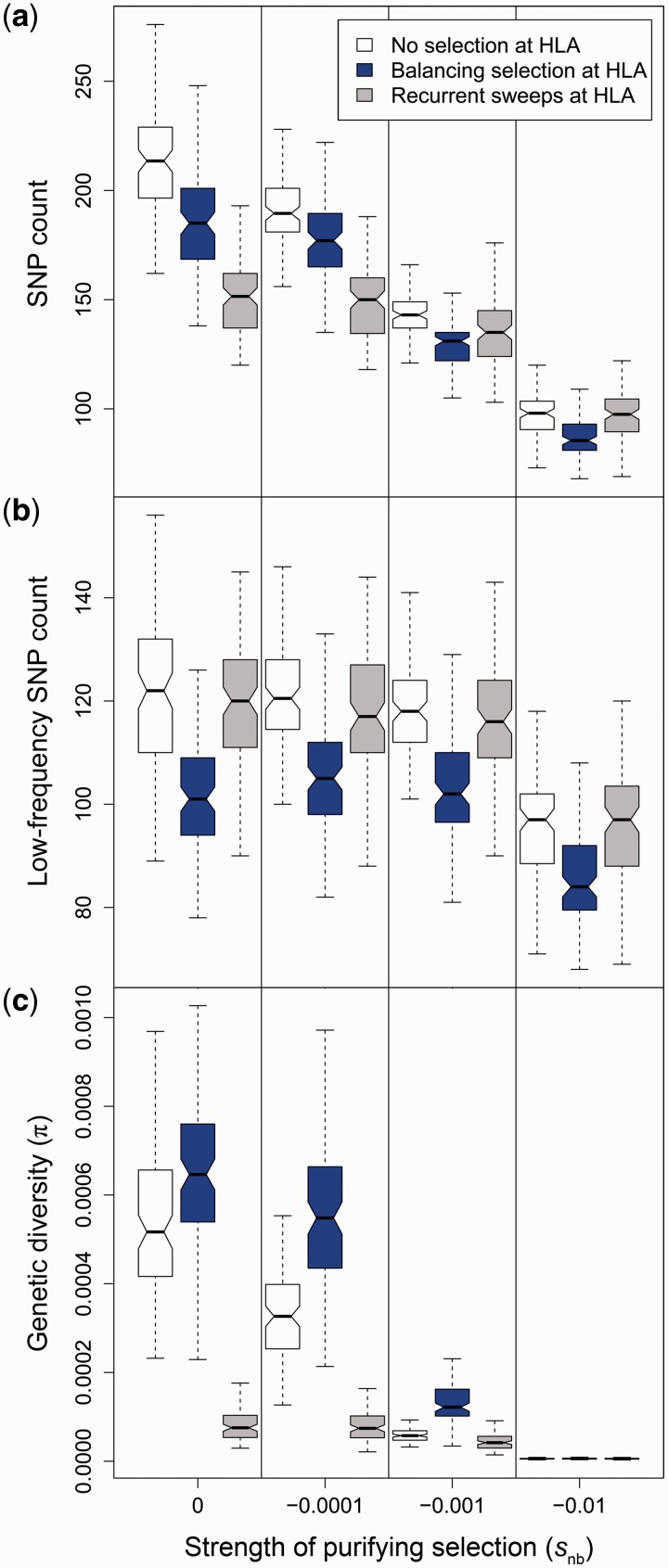
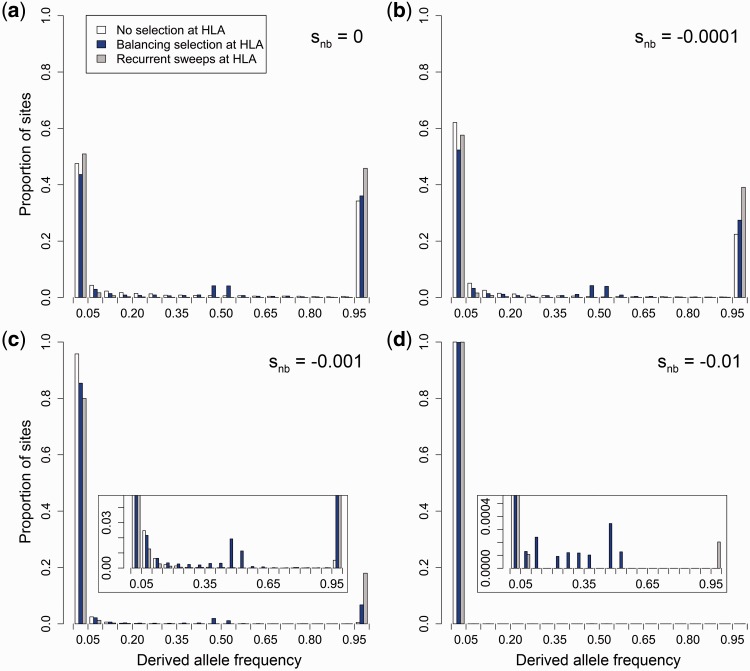
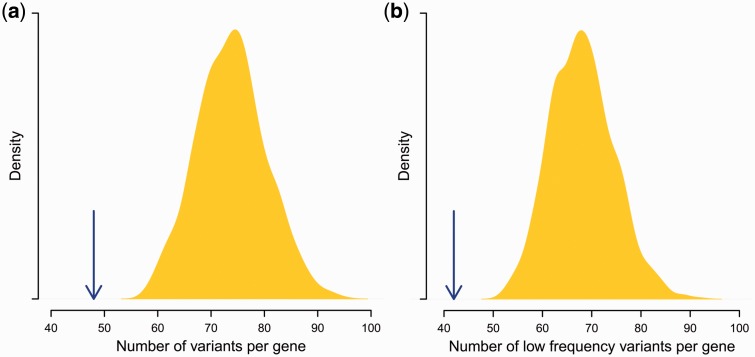
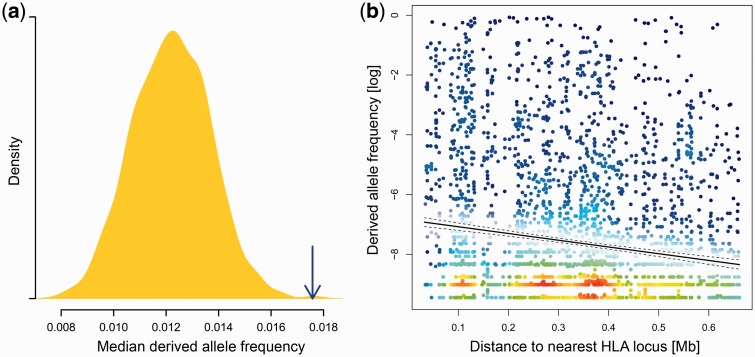
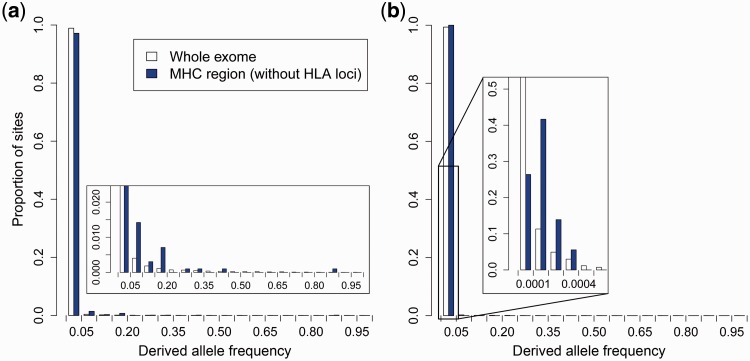
Deleterious mutations are expected to evolve under negative selection and are usually purged from the population. However, deleterious alleles segregate in the human population and some disease-associated variants are maintained at considerable frequencies. Here, we test the hypothesis that balancing selection may counteract purifying selection in neighboring regions and thus maintain deleterious variants at higher frequency than expected from their detrimental fitness effect. We first show in realistic simulations that balancing selection reduces the density of polymorphic sites surrounding a locus under balancing selection, but at the same time markedly increases the population frequency of the remaining variants, including even substantially deleterious alleles. To test the predictions of our simulations empirically, we then use whole-exome sequencing data from 6,500 human individuals and focus on the most established example for balancing selection in the human genome, the major histocompatibility complex (MHC). Our analysis shows an elevated frequency of putatively deleterious coding variants in nonhuman leukocyte antigen (non-HLA) genes localized in the MHC region. The mean frequency of these variants declined with physical distance from the classical HLA genes, indicating dependency on genetic linkage. These results reveal an indirect cost of the genetic diversity maintained by balancing selection, which has hitherto been perceived as mostly advantageous, and have implications both for the evolution of recombination and also for the epidemiology of various MHC-associated diseases.
Keywords: MHC/HLA.; balancing selection; deleterious variation; exome; mutation load; simulations.
© The Author 2016. Published by Oxford University Press on behalf of the Society for Molecular Biology and Evolution.
Figures
Comment in
-
Lives in the Balance: Why Do We Hold onto Potentially Harmful, Disease-Causing Mutations?Mol Biol Evol. 2016 Oct;33(10):2765-6. doi: 10.1093/molbev/msw146. Epub 2016 Aug 30. Mol Biol Evol. 2016. PMID: 27582520 No abstract available.
References
-
- Asthana S, Schmidt S, Sunyaev SR. 2005. A limited role for balancing selection. Trends Genet. 21:30–32. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
