

Indel Group in Genomes (IGG) Molecular Genetic Markers
- PMID: 27436831
- PMCID: PMC5074621
- DOI: 10.1104/pp.16.00354
Indel Group in Genomes (IGG) Molecular Genetic Markers
Abstract
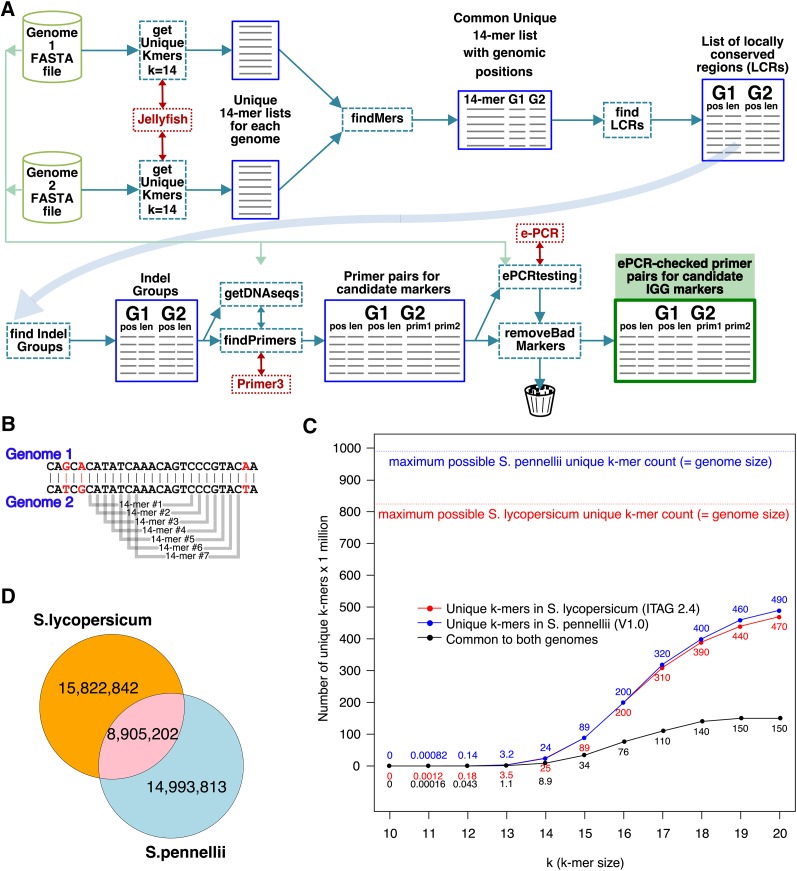
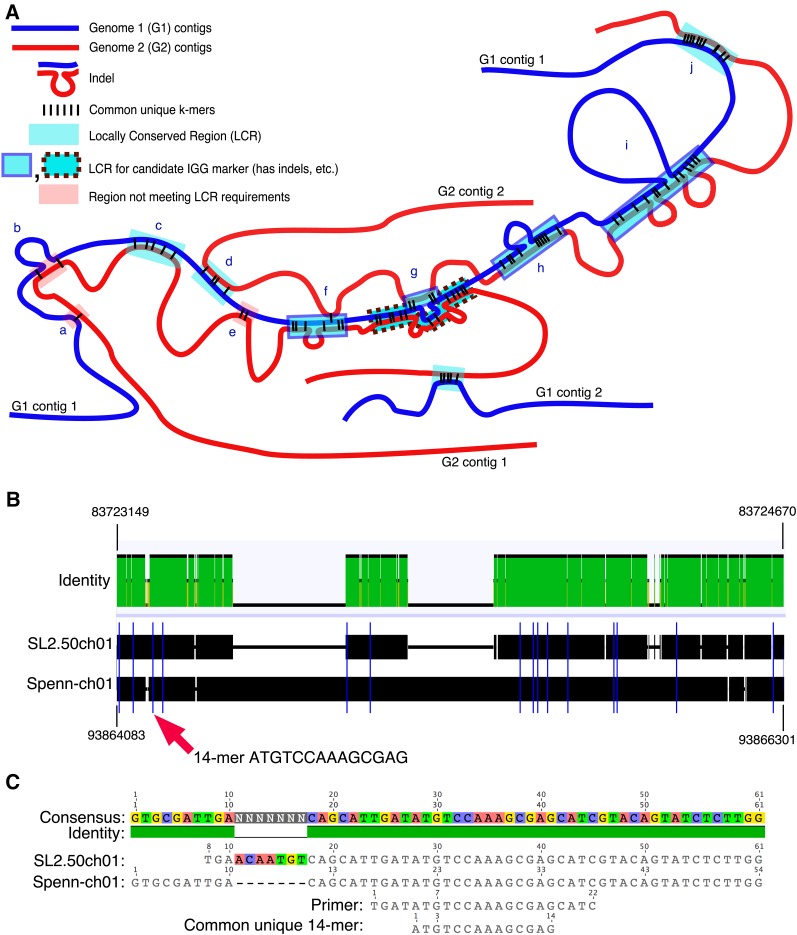
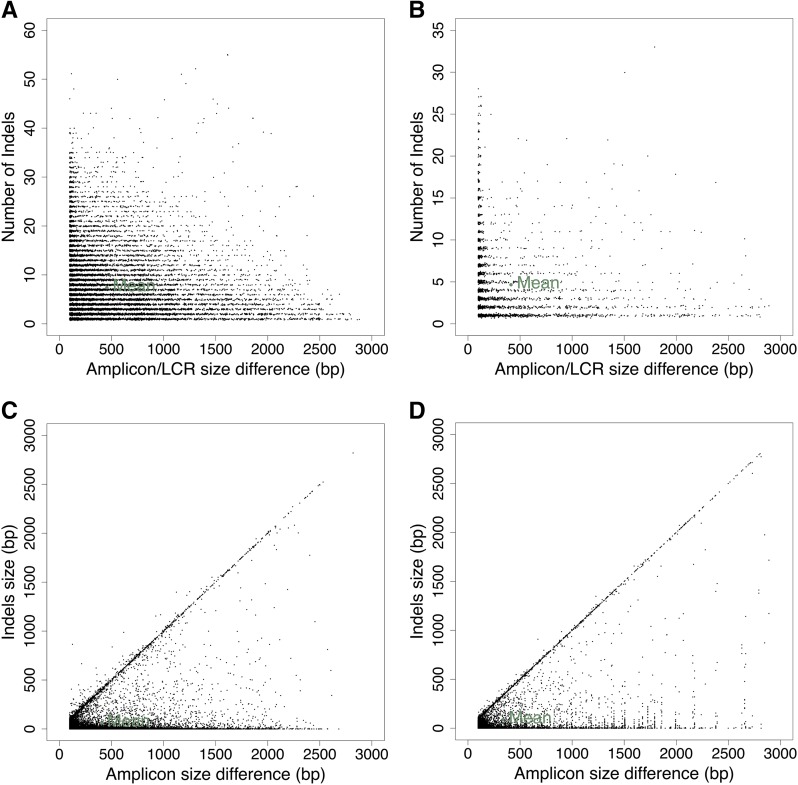
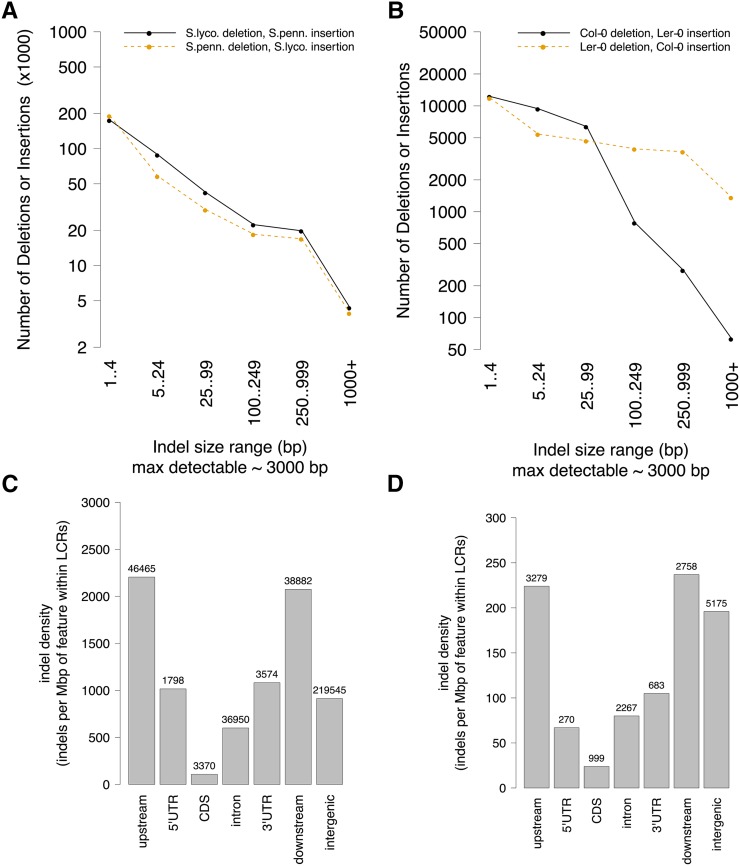
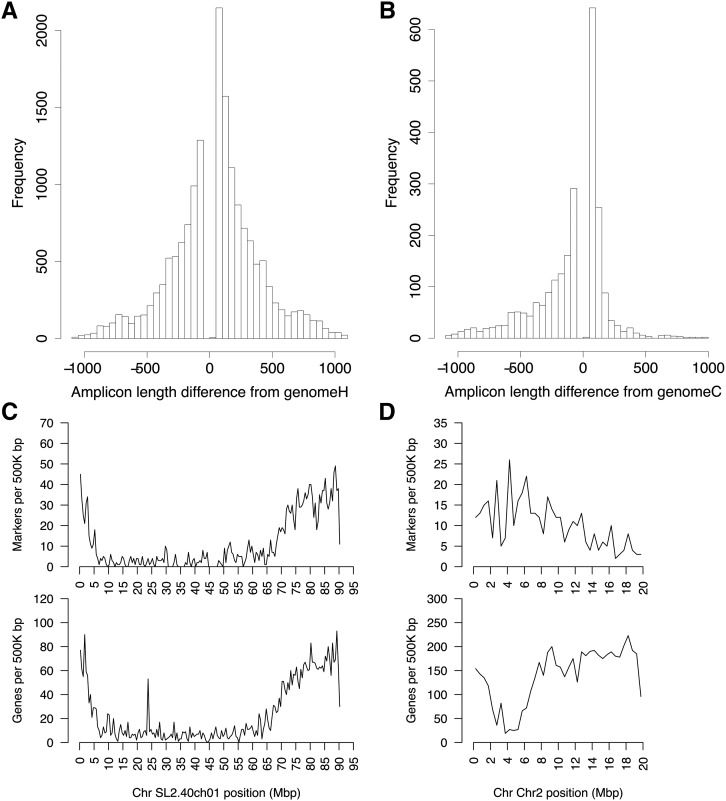
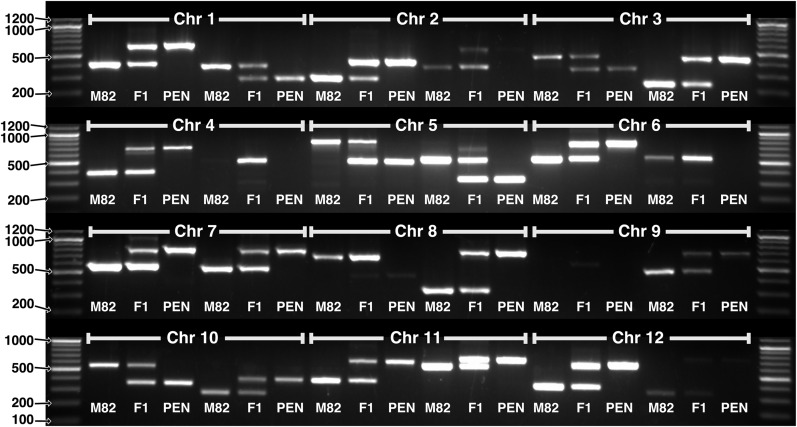
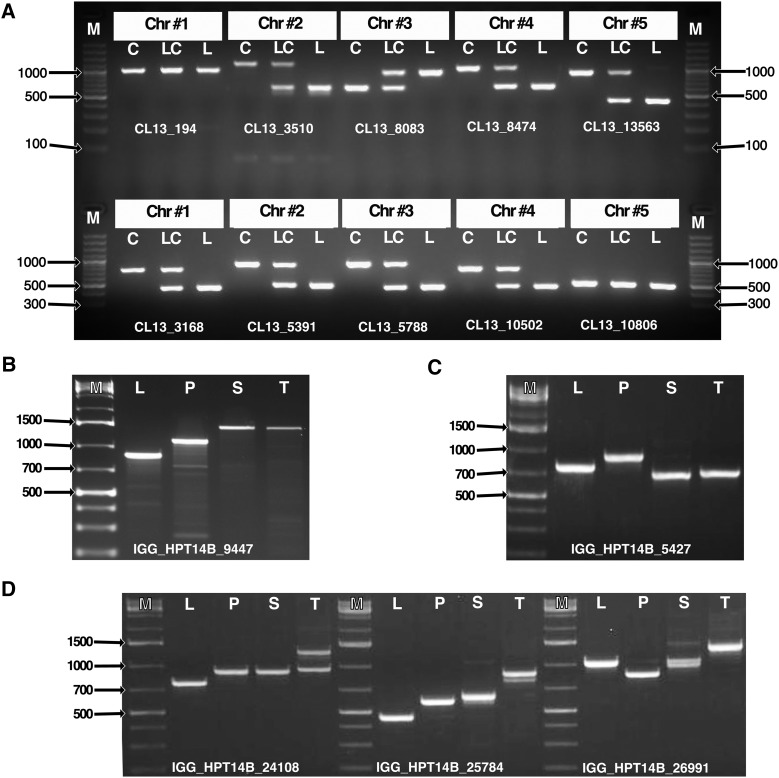
Genetic markers are essential when developing or working with genetically variable populations. Indel Group in Genomes (IGG) markers are primer pairs that amplify single-locus sequences that differ in size for two or more alleles. They are attractive for their ease of use for rapid genotyping and their codominant nature. Here, we describe a heuristic algorithm that uses a k-mer-based approach to search two or more genome sequences to locate polymorphic regions suitable for designing candidate IGG marker primers. As input to the IGG pipeline software, the user provides genome sequences and the desired amplicon sizes and size differences. Primer sequences flanking polymorphic insertions/deletions are produced as output. IGG marker files for three sets of genomes, Solanum lycopersicum/Solanum pennellii, Arabidopsis (Arabidopsis thaliana) Columbia-0/Landsberg erecta-0 accessions, and S. lycopersicum/S. pennellii/Solanum tuberosum (three-way polymorphic) are included.
© 2016 American Society of Plant Biologists. All rights reserved.
Figures
Similar articles
-
Evidence of cryptic introgression in tomato (Solanum lycopersicum L.) based on wild tomato species alleles.BMC Plant Biol. 2012 Aug 7;12:133. doi: 10.1186/1471-2229-12-133. BMC Plant Biol. 2012. PMID: 22871151 Free PMC article.
-
In silico identification and experimental validation of insertion-deletion polymorphisms in tomato genome.DNA Res. 2014 Aug;21(4):429-38. doi: 10.1093/dnares/dsu008. Epub 2014 Mar 11. DNA Res. 2014. PMID: 24618211 Free PMC article.
-
Development of a set of PCR-based anchor markers encompassing the tomato genome and evaluation of their usefulness for genetics and breeding experiments.Theor Appl Genet. 2005 Jul;111(2):291-312. doi: 10.1007/s00122-005-2023-7. Epub 2005 May 31. Theor Appl Genet. 2005. PMID: 15926074
-
Bin mapping of tomato diversity array (DArT) markers to genomic regions of Solanum lycopersicum × Solanum pennellii introgression lines.Theor Appl Genet. 2012 Mar;124(5):947-56. doi: 10.1007/s00122-011-1759-5. Epub 2011 Dec 13. Theor Appl Genet. 2012. PMID: 22159755 Free PMC article.
-
Mapping of BnMs4 and BnRf to a common microsyntenic region of Arabidopsis thaliana chromosome 3 using intron polymorphism markers.Theor Appl Genet. 2012 May;124(7):1193-200. doi: 10.1007/s00122-011-1779-1. Epub 2012 Jan 14. Theor Appl Genet. 2012. PMID: 22246313 Review.
Cited by
-
Never the Two Shall Mix: Robust Indel Markers to Ensure the Fidelity of Two Pivotal and Closely-Related Accessions of Brachypodium distachyon.Plants (Basel). 2019 Jun 6;8(6):153. doi: 10.3390/plants8060153. Plants (Basel). 2019. PMID: 31174296 Free PMC article.
References
-
- Ahmed A, Ferreira AS, Hartskeerl RA (2015) Multilocus sequence typing (MLST): markers for the traceability of pathogenic Leptospira strains. Methods Mol Biol 1247: 349–359 - PubMed
-
- Bombarely A, Menda N, Tecle IY, Buels RM, Strickler S, Fischer-York T, Pujar A, Leto J, Gosselin J, Mueller LA (2011) The Sol Genomics Network (solgenomics.net): growing tomatoes using Perl. Nucleic Acids Res 39: D1149–D1155 - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous
