

Transcriptional control of amino acid homeostasis is disrupted in Huntington's disease
- PMID: 27436896
- PMCID: PMC4978294
- DOI: 10.1073/pnas.1608264113
Transcriptional control of amino acid homeostasis is disrupted in Huntington's disease
Abstract
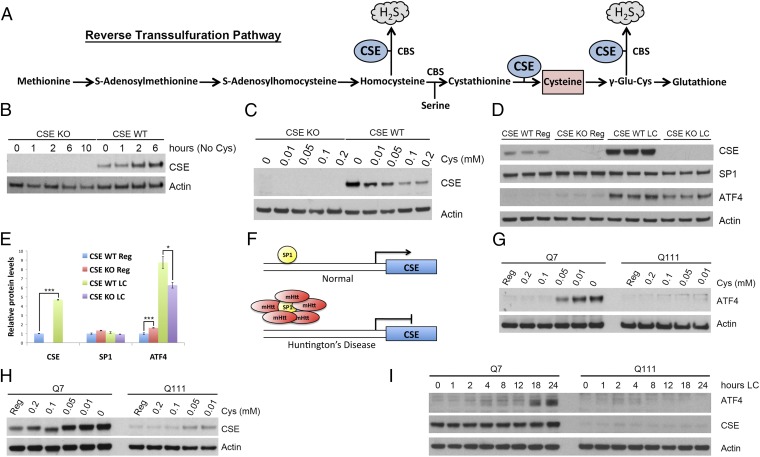
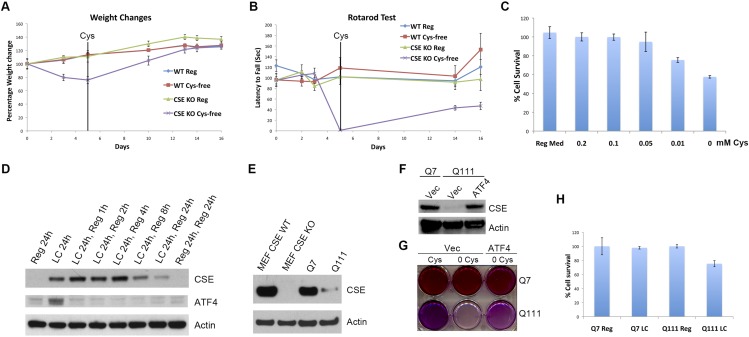
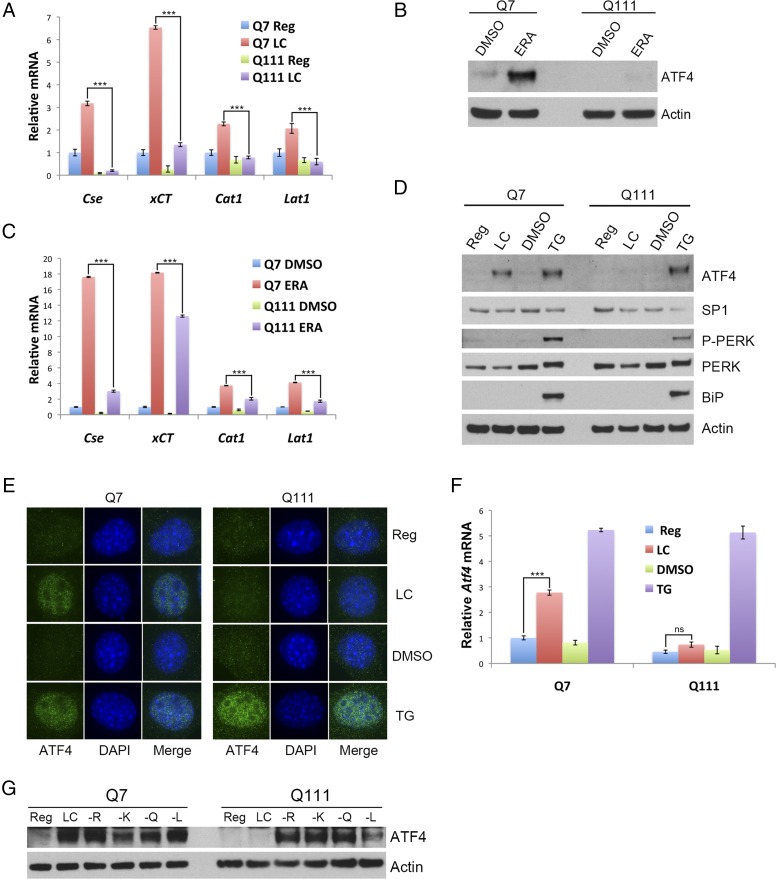
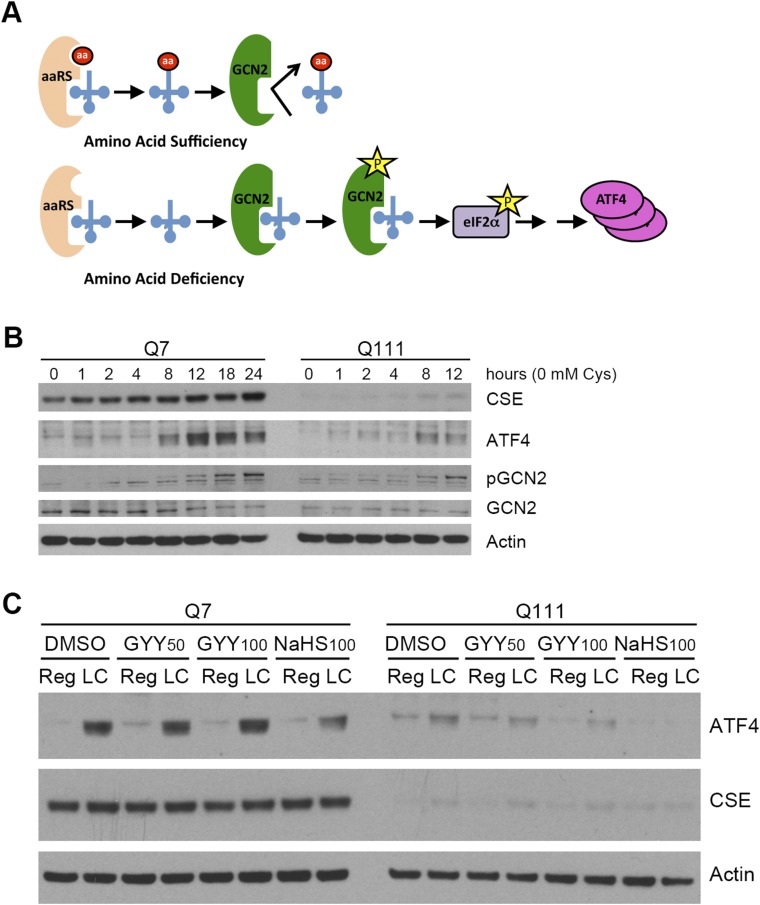
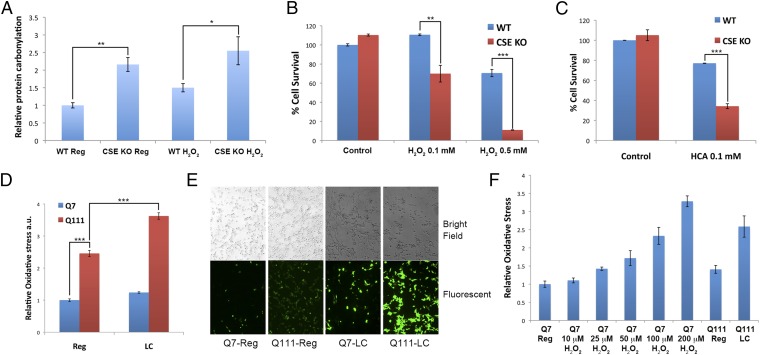
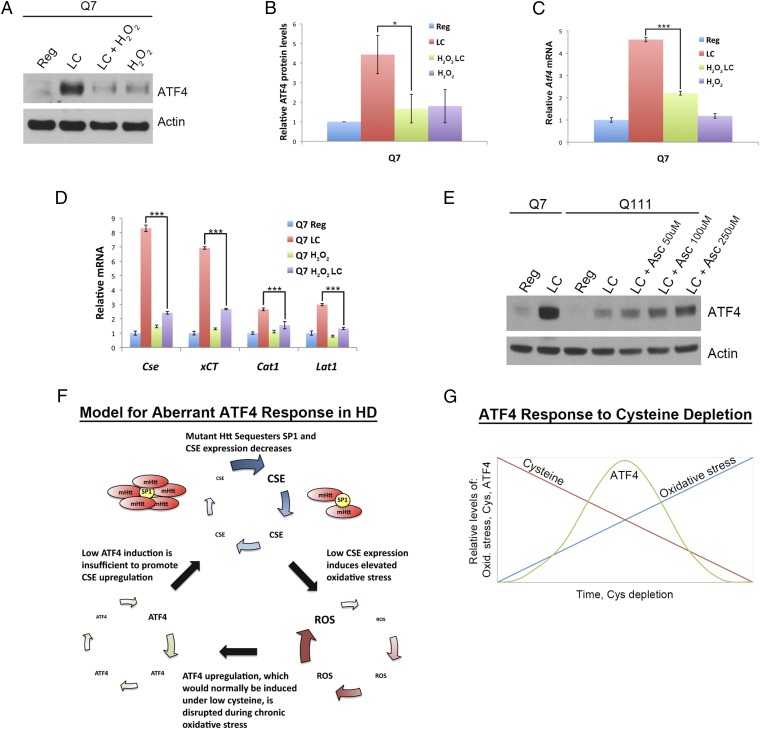
Disturbances in amino acid metabolism, which have been observed in Huntington's disease (HD), may account for the profound inanition of HD patients. HD is triggered by an expansion of polyglutamine repeats in the protein huntingtin (Htt), impacting diverse cellular processes, ranging from transcriptional regulation to cognitive and motor functions. We show here that the master regulator of amino acid homeostasis, activating transcription factor 4 (ATF4), is dysfunctional in HD because of oxidative stress contributed by aberrant cysteine biosynthesis and transport. Consistent with these observations, antioxidant supplementation reverses the disordered ATF4 response to nutrient stress. Our findings establish a molecular link between amino acid disposition and oxidative stress leading to cytotoxicity. This signaling cascade may be relevant to other diseases involving redox imbalance and deficits in amino acid metabolism.
Keywords: ATF4; CSE; Huntington’s disease; cysteine; oxidative stress.
Conflict of interest statement
The authors declare no conflict of interest.
Figures
Similar articles
-
Impaired Redox Signaling in Huntington's Disease: Therapeutic Implications.Front Mol Neurosci. 2019 Mar 19;12:68. doi: 10.3389/fnmol.2019.00068. eCollection 2019. Front Mol Neurosci. 2019. PMID: 30941013 Free PMC article. Review.
-
Golgi stress response reprograms cysteine metabolism to confer cytoprotection in Huntington's disease.Proc Natl Acad Sci U S A. 2018 Jan 23;115(4):780-785. doi: 10.1073/pnas.1717877115. Epub 2018 Jan 9. Proc Natl Acad Sci U S A. 2018. PMID: 29317536 Free PMC article.
-
Cystathionine γ-lyase deficiency mediates neurodegeneration in Huntington's disease.Nature. 2014 May 1;509(7498):96-100. doi: 10.1038/nature13136. Epub 2014 Mar 26. Nature. 2014. PMID: 24670645 Free PMC article.
-
Cysteine metabolism and hydrogen sulfide signaling in Huntington's disease.Free Radic Biol Med. 2022 Jun;186:93-98. doi: 10.1016/j.freeradbiomed.2022.05.005. Epub 2022 May 10. Free Radic Biol Med. 2022. PMID: 35550919 Free PMC article. Review.
-
Integrated stress response modulates cellular redox state via induction of cystathionine γ-lyase: cross-talk between integrated stress response and thiol metabolism.J Biol Chem. 2012 Mar 2;287(10):7603-14. doi: 10.1074/jbc.M111.304576. Epub 2012 Jan 3. J Biol Chem. 2012. PMID: 22215680 Free PMC article.
Cited by
-
Hydrogen sulfide signaling in mitochondria and disease.FASEB J. 2019 Dec;33(12):13098-13125. doi: 10.1096/fj.201901304R. Epub 2019 Oct 24. FASEB J. 2019. PMID: 31648556 Free PMC article. Review.
-
Integrative Analysis Unveils the Correlation of Aminoacyl-tRNA Biosynthesis Metabolites with the Methylation of the SEPSECS Gene in Huntington's Disease Brain Tissue.Genes (Basel). 2023 Sep 2;14(9):1752. doi: 10.3390/genes14091752. Genes (Basel). 2023. PMID: 37761892 Free PMC article.
-
Golgi Complex Dynamics and Its Implication in Prevalent Neurological Disorders.Front Cell Dev Biol. 2019 May 7;7:75. doi: 10.3389/fcell.2019.00075. eCollection 2019. Front Cell Dev Biol. 2019. PMID: 31134199 Free PMC article. Review.
-
Impaired Redox Signaling in Huntington's Disease: Therapeutic Implications.Front Mol Neurosci. 2019 Mar 19;12:68. doi: 10.3389/fnmol.2019.00068. eCollection 2019. Front Mol Neurosci. 2019. PMID: 30941013 Free PMC article. Review.
-
Hydrogen sulfide is neuroprotective in Alzheimer's disease by sulfhydrating GSK3β and inhibiting Tau hyperphosphorylation.Proc Natl Acad Sci U S A. 2021 Jan 26;118(4):e2017225118. doi: 10.1073/pnas.2017225118. Proc Natl Acad Sci U S A. 2021. PMID: 33431651 Free PMC article.
References
-
- The Huntington’s Disease Collaborative Research Group A novel gene containing a trinucleotide repeat that is expanded and unstable on Huntington’s disease chromosomes. Cell. 1993;72(6):971–983. - PubMed
-
- Davies SW, et al. Formation of neuronal intranuclear inclusions underlies the neurological dysfunction in mice transgenic for the HD mutation. Cell. 1997;90(3):537–548. - PubMed
-
- Sugars KL, Rubinsztein DC. Transcriptional abnormalities in Huntington disease. Trends Genet. 2003;19(5):233–238. - PubMed
-
- Zhai W, Jeong H, Cui L, Krainc D, Tjian R. In vitro analysis of huntingtin-mediated transcriptional repression reveals multiple transcription factor targets. Cell. 2005;123(7):1241–1253. - PubMed
-
- Mochel F, Benaich S, Rabier D, Durr A. Validation of plasma branched chain amino acids as biomarkers in Huntington disease. Arch Neurol. 2011;68(2):265–267. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
