

Effects of fingolimod administration in a genetic model of cognitive deficits
- PMID: 27439747
- PMCID: PMC6636312
- DOI: 10.1002/jnr.23799
Effects of fingolimod administration in a genetic model of cognitive deficits
Abstract
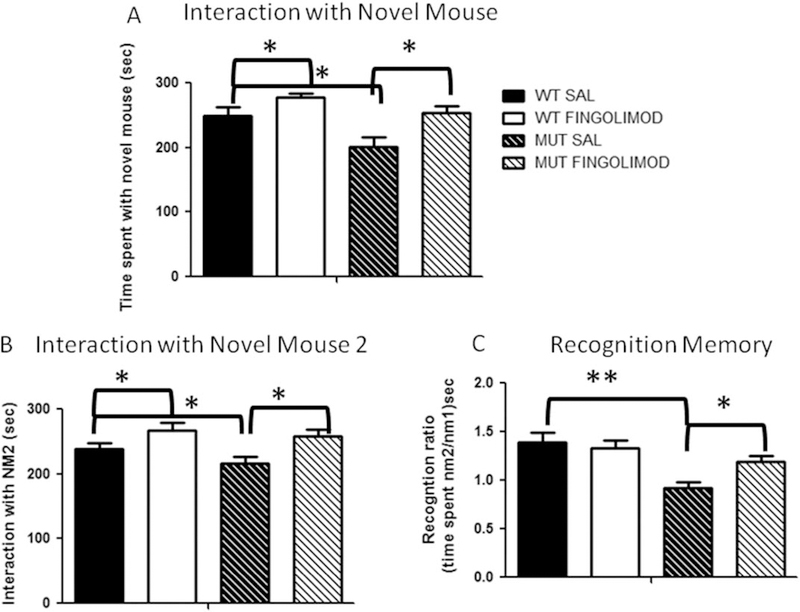
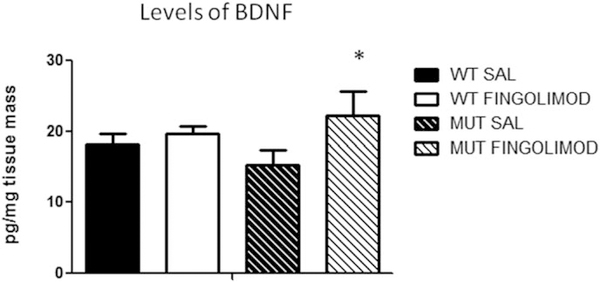
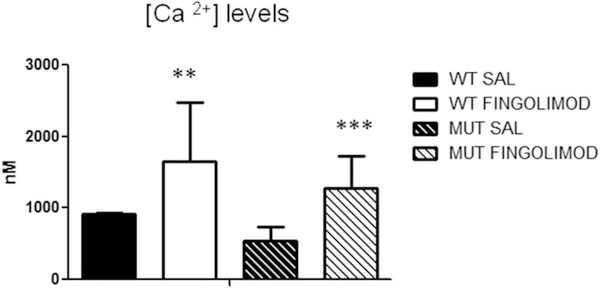
Notwithstanding recent advances, cognitive impairments are among the most difficult-to-treat symptoms in neuropsychiatric disorders. Deficits in information processing contributing to memory and sociability impairments are found across neuropsychiatric-related disorders. Previously, we have shown that mutations in the DTNBP1 gene (encoding dystrobrevin-binding protein 1 [dysbindin-1]) lead to abnormalities in synaptic glutamate release in the prefrontal cortex (PFC) and hippocampus and to cognitive deficits; glutamatergic transmission is important for cortical recurrent excitation that allows information processing in the PFC. To investigate possible means of restoring glutamate release and improving cognitive impairments, we assess the effects of increasing endogenous levels of brain-derived neurotrophic factor (BDNF) in a dysbindin-1-deficient mouse model. Increasing endogenous levels of BDNF may aid in remediating cognitive deficits, given the roles of BDNF in synaptic transmission, plasticity, and neuroprotection. To increase BDNF, we use a novel strategy, repeated intraperitoneal injections of fingolimod (Gilenya). Sphingolipids have recently been shown to have therapeutic value in several neurology-related disorders. Both wild-type (WT) and mutant (MUT) genotypes were tested for sociability and recognition memory, followed by measuring endogenous BDNF levels and presynaptic [Ca2+ ]i within the PFC. Both genotypes were treated for 1 week with either saline or fingolimod. Relative to WT mice, MUT mice demonstrated impairments in sociability and recognition memory and lower presynaptic calcium. After fingolimod treatment, MUT mice exhibited significant improvements in sociability and recognition memory and increases in presynaptic calcium and endogenous concentrations of BDNF. These results show promise for counteracting the cognitive impairments seen in neuropsychiatric disorders and may shed light on the role of dysbindin-1. © 2016 Wiley Periodicals, Inc.
Keywords: dysbindin; fingolimod; glutamate; prefrontal cortex.
© 2016 Wiley Periodicals, Inc.
Conflict of interest statement
CONFLICT OF INTEREST STATEMENT
The authors do not have any conflicts of interest.
Figures
References
-
- Baldelli P, Forni PE, Carbone E. 2000. BDNF, NT-3, and NGF induce distinct new Ca2+ channel synthesis in developing hippocampal neurons. Eur J Neurosci 12:4017–4032. - PubMed
-
- Baldelli P, Novara M, Carabelli V, Hernández-Guijo JM, Carbone E. 2002. BDNF upregulates evoked GABAergic transmission in developing hippocampus by potentiating presynaptic N- and P/Q-type Ca2+ channels signalling. Eur J Neurosci 16:2297–2310. - PubMed
-
- Berridge MJ. 1998. Neuronal calcium signaling. Neuron 21:13–26. - PubMed
-
- Callicott JH, Bertolino A, Mattay VS, Langheim FJ, Duyn J, Coppola R, Goldberg TE, Weinberger DR. 2000. Physiological dysfunction of the dorsolateral prefrontal cortex in schizophrenia revisited. Cereb Cortex 10:1078–1092. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
