

AMP-Activated Protein Kinase Alpha 2 Deletion Induces VSMC Phenotypic Switching and Reduces Features of Atherosclerotic Plaque Stability
- PMID: 27439892
- PMCID: PMC6265658
- DOI: 10.1161/CIRCRESAHA.116.308689
AMP-Activated Protein Kinase Alpha 2 Deletion Induces VSMC Phenotypic Switching and Reduces Features of Atherosclerotic Plaque Stability
Abstract
Rationale: AMP-activated protein kinase (AMPK) has been reported to play a protective role in atherosclerosis. However, whether AMPKα2 controls atherosclerotic plaque stability remains unknown.
Objective: The aim of this study was to evaluate the impact of AMPKα2 deletion on atherosclerotic plaque stability in advanced atherosclerosis at the brachiocephalic arteries and to elucidate the underlying mechanisms.
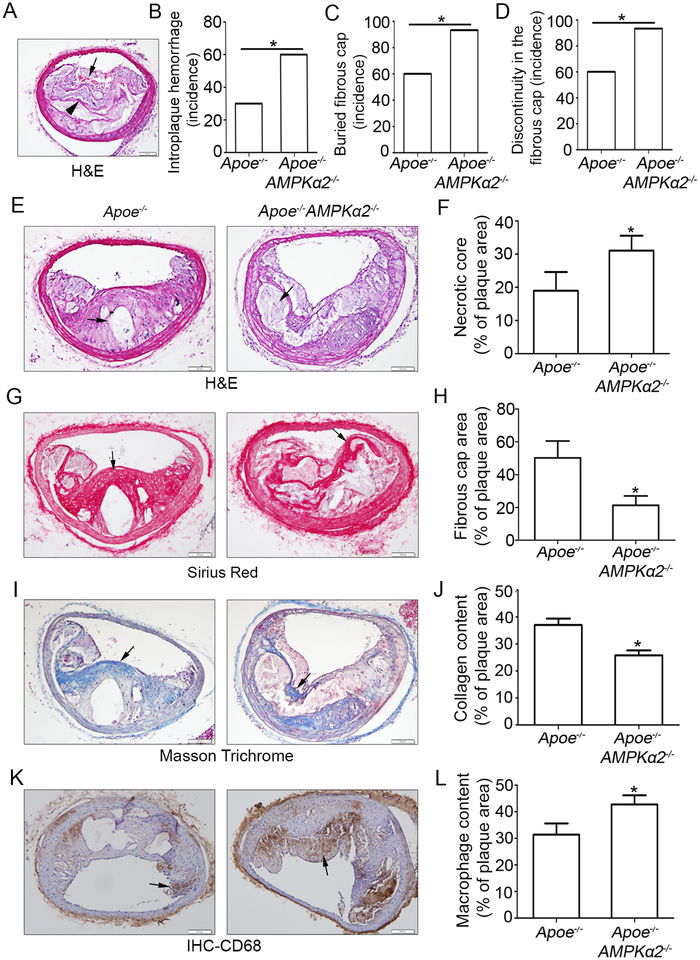
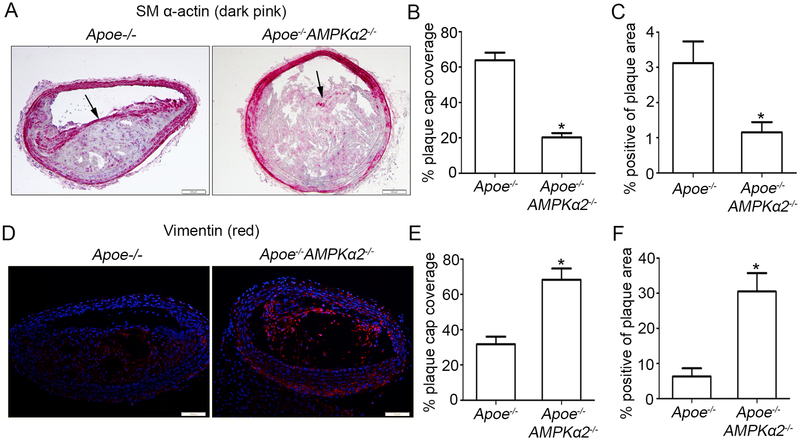
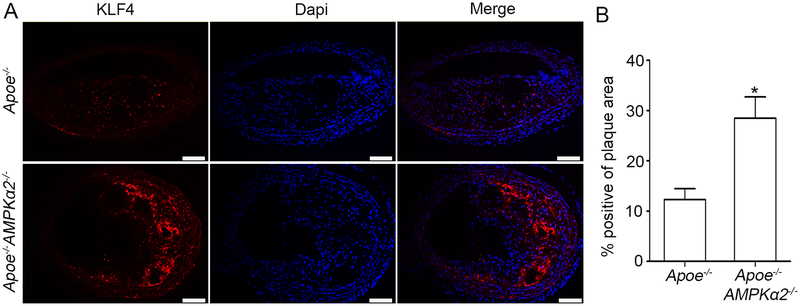
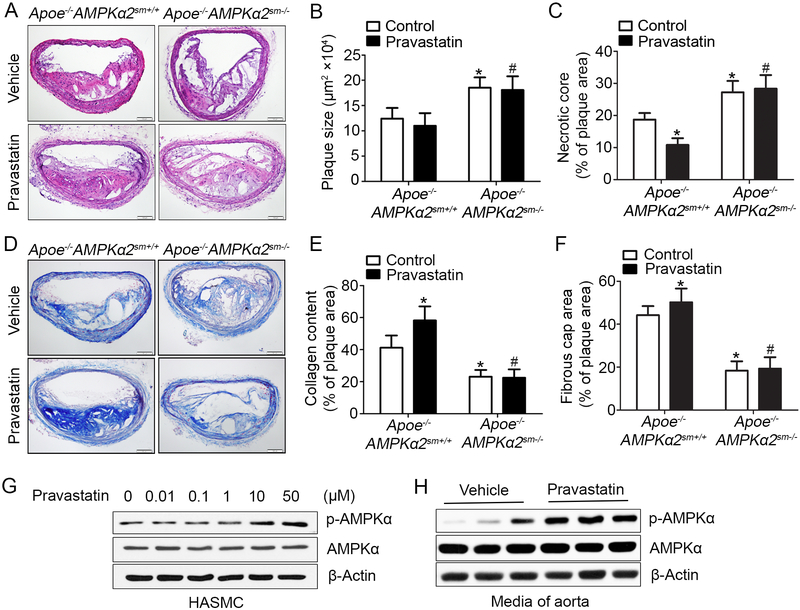
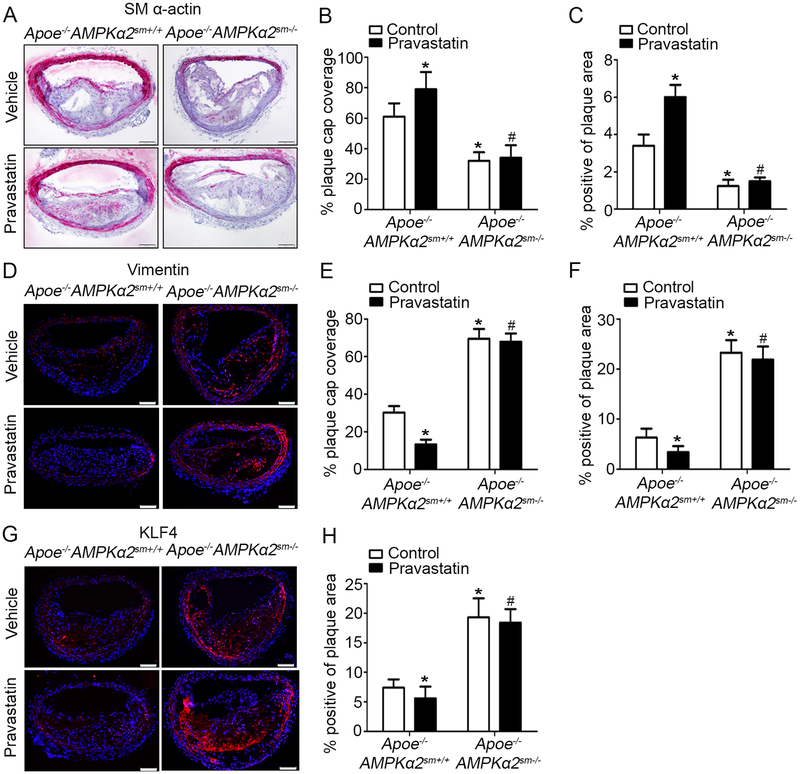
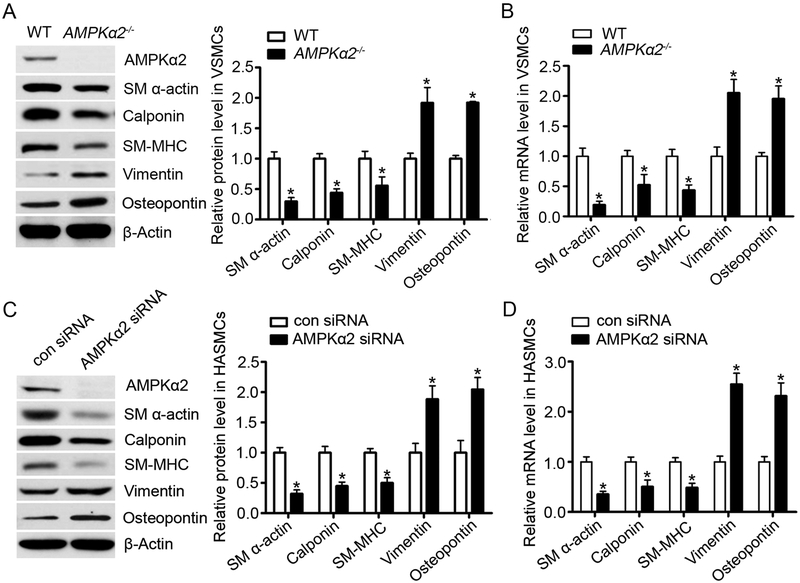
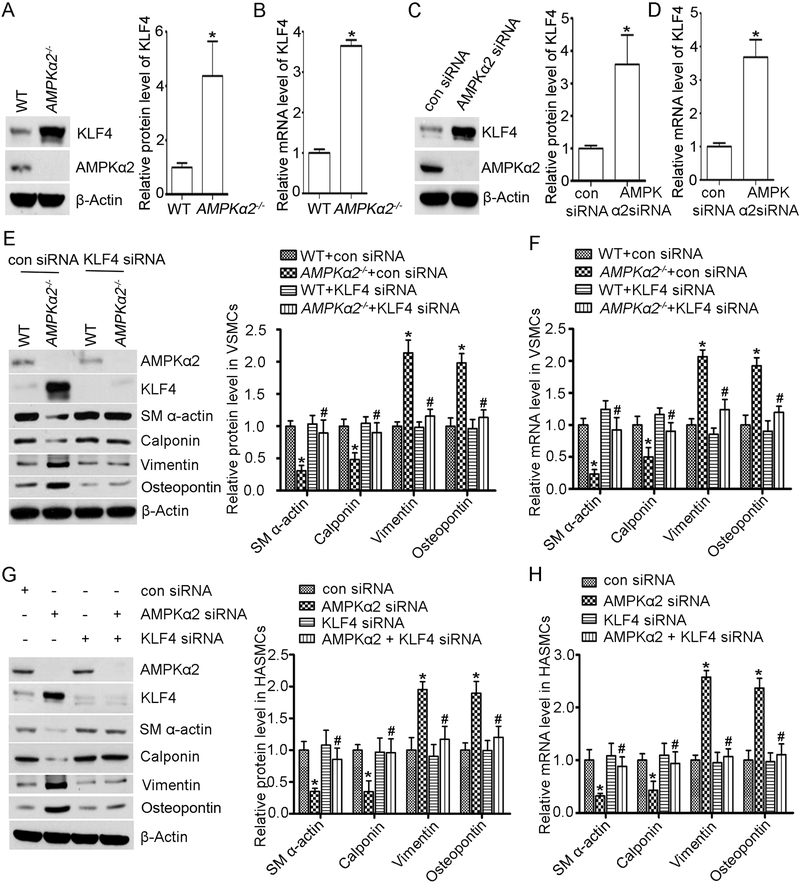
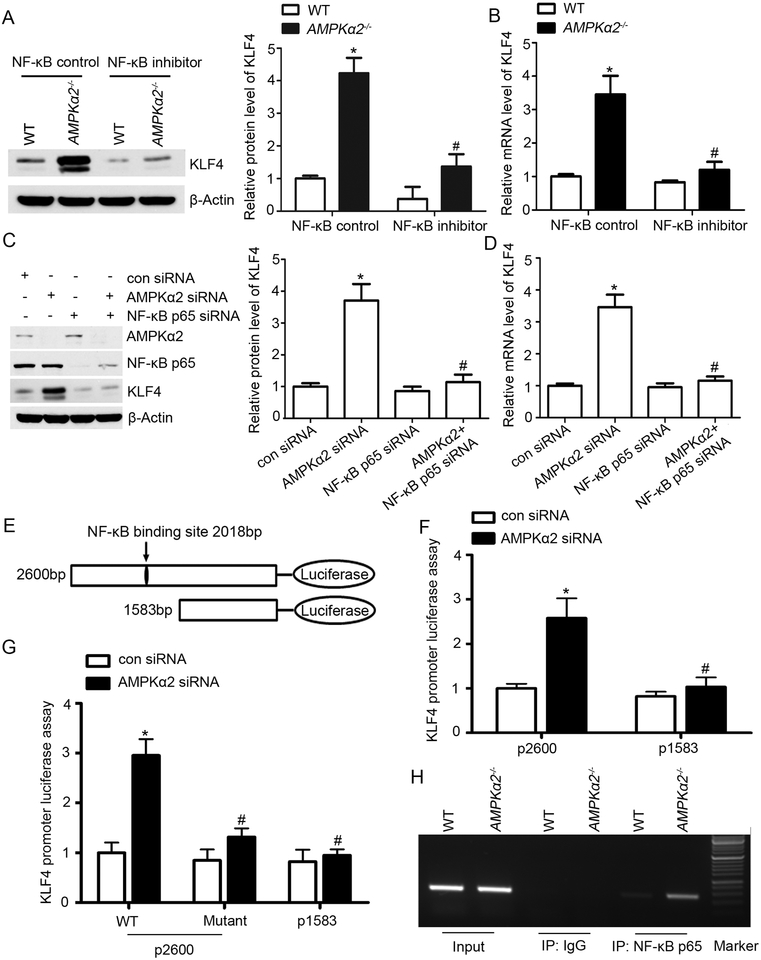
Methods and results: Features of atherosclerotic plaque stability and the markers for contractile or synthetic vascular smooth muscle cell (VSMC) phenotypes were monitored in the brachiocephalic arteries from Apoe(-/-)AMPKα2(-/-) mice or VSMC-specific AMPKα2(-/-) mice in an Apoe(-/-) background (Apoe(-/-)AMPKα2(sm-/-)) fed Western diet for 10 weeks. We identified that Apoe(-/-)AMPKα2(-/-) mice and Apoe(-/-)AMPKα2(sm-/-) mice exhibited similar unstable plaque features, aggravated VSMC phenotypic switching, and significant upregulation of Kruppel-like factor 4 (KLF4) in the plaques located in the brachiocephalic arteries compared with those found in Apoe(-/-) and Apoe(-/-)AMPKα2(sm+/+) control mice. Pravastatin, an AMPK activator, suppressed VSMC phenotypic switching and alleviated features of atherosclerotic plaque instability in Apoe(-/-)AMPKα2(sm+/+) mice, but not in Apoe(-/-)AMPKα2(sm-/-) mice. VSMC isolated from AMPKα2(-/-) mice displayed a significant reduction of contractile proteins(smooth muscle actin-α, calponin, and SM-MHC [smooth muscle-mysion heavy chain]) in parallel with increased detection of synthetic proteins (vimentin and osteopontin) and KLF4, as observed in vivo. KLF4-specific siRNA abolished AMPKα2 deletion-induced VSMC phenotypic switching. Furthermore, pharmacological or genetic inhibition of nuclear factor-κB significantly decreased KLF4 upregulation in VSMC from AMPKα2(-/-) mice. Finally, we found that AMPKα2 deletion markedly promoted the binding of nuclear factor-κBp65 to KLF4 promoter.
Conclusions: This study demonstrated that AMPKα2 deletion induces VSMC phenotypic switching and promotes features of atherosclerotic plaque instability in nuclear factor-κB-KLF4-dependent manner.
Keywords: AMP-activated protein kinases; atherosclerosis.
© 2016 American Heart Association, Inc.
Figures
References
-
- Hansson GK. Inflammation, atherosclerosis, and coronary artery disease. N Engl J Med. 2005;352:1685–1695 - PubMed
-
- Yla-Herttuala S, Bentzon JF, Daemen M, Falk E, Garcia-Garcia HM, Herrmann J, Hoefer I, Jukema JW, Krams R, Kwak BR, Marx N, Naruszewicz M, Newby A, Pasterkamp G, Serruys PW, Waltenberger J, Weber C, Tokgozoglu L. Stabilisation of atherosclerotic plaques. Position paper of the european society of cardiology (esc) working group on atherosclerosis and vascular biology. Thromb Haemost. 2011;106:1–19 - PubMed
-
- Rzucidlo EM, Martin KA, Powell RJ. Regulation of vascular smooth muscle cell differentiation. J Vasc Surg. 2007;45 Suppl A:A25–32 - PubMed
-
- Bennett MR. Apoptosis of vascular smooth muscle cells in vascular remodelling and atherosclerotic plaque rupture. Cardiovasc Res. 1999;41:361–368 - PubMed
MeSH terms
Substances
Grants and funding
- R01 HL089920/HL/NHLBI NIH HHS/United States
- R01 CA213022/CA/NCI NIH HHS/United States
- R01 HL128014/HL/NHLBI NIH HHS/United States
- R01 HL074399/HL/NHLBI NIH HHS/United States
- R01 AG047776/AG/NIA NIH HHS/United States
- R01 HL137371/HL/NHLBI NIH HHS/United States
- R01 HL110488/HL/NHLBI NIH HHS/United States
- R01 HL105157/HL/NHLBI NIH HHS/United States
- R01 HL142287/HL/NHLBI NIH HHS/United States
- R01 HL079584/HL/NHLBI NIH HHS/United States
- R01 HL140954/HL/NHLBI NIH HHS/United States
- R01 HL080499/HL/NHLBI NIH HHS/United States
- R01 HL096032/HL/NHLBI NIH HHS/United States
- R01 HL132500/HL/NHLBI NIH HHS/United States
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Research Materials
Miscellaneous
