

Genetics of complex traits: prediction of phenotype, identification of causal polymorphisms and genetic architecture
- PMID: 27440663
- PMCID: PMC4971198
- DOI: 10.1098/rspb.2016.0569
Genetics of complex traits: prediction of phenotype, identification of causal polymorphisms and genetic architecture
Abstract
Complex or quantitative traits are important in medicine, agriculture and evolution, yet, until recently, few of the polymorphisms that cause variation in these traits were known. Genome-wide association studies (GWAS), based on the ability to assay thousands of single nucleotide polymorphisms (SNPs), have revolutionized our understanding of the genetics of complex traits. We advocate the analysis of GWAS data by a statistical method that fits all SNP effects simultaneously, assuming that these effects are drawn from a prior distribution. We illustrate how this method can be used to predict future phenotypes, to map and identify the causal mutations, and to study the genetic architecture of complex traits. The genetic architecture of complex traits is even more complex than previously thought: in almost every trait studied there are thousands of polymorphisms that explain genetic variation. Methods of predicting future phenotypes, collectively known as genomic selection or genomic prediction, have been widely adopted in livestock and crop breeding, leading to increased rates of genetic improvement.
Keywords: complex traits; genome-wide association studies; genomic selection.
© 2016 The Author(s).
Figures
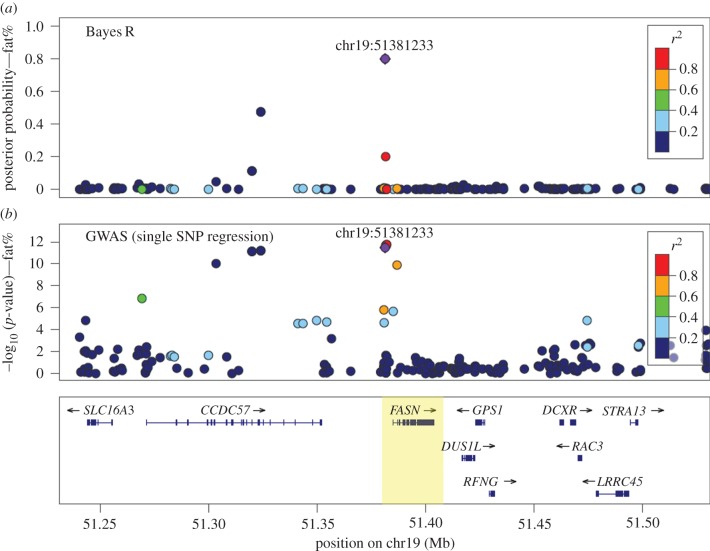
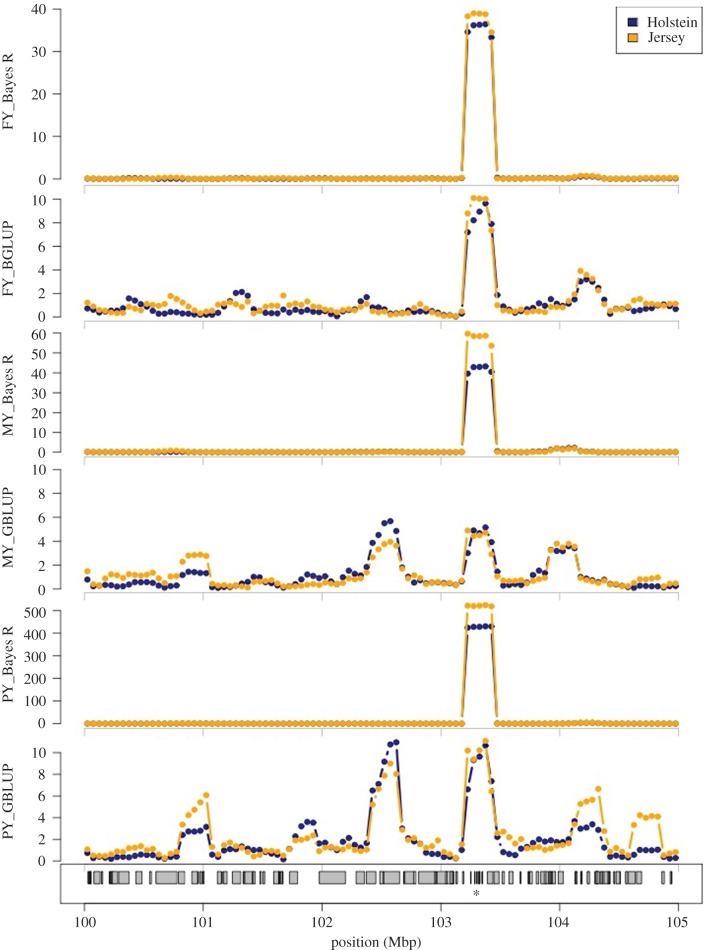
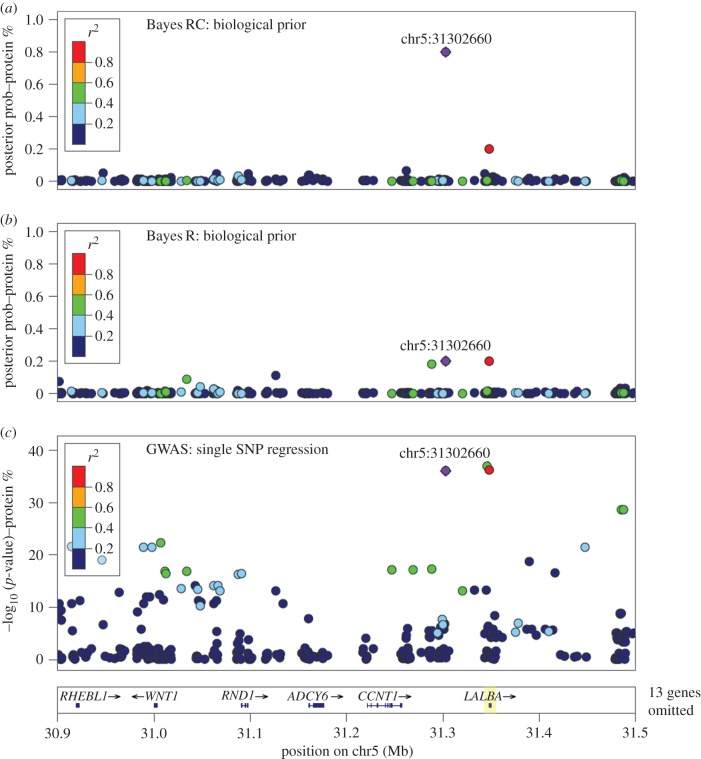
References
-
- Goddard ME, Wray NR, Verbyla K, Visscher PM. 2009. Estimating effects and making predictions from genome-wide marker data. Stat. Sci. 24, 517–529. (10.1214/09-STS306) - DOI
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
