

Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice
- PMID: 27440721
- PMCID: PMC5199750
- DOI: 10.1113/JP272950
Progressive impairment of cerebellar mGluR signalling and its therapeutic potential for cerebellar ataxia in spinocerebellar ataxia type 1 model mice
Abstract
Key points: Spinocerebellar ataxia type 1 (SCA1) is a progressive neurodegenerative disease caused by a gene defect, leading to movement disorder such as cerebellar ataxia. It remains largely unknown which functional defect contributes to the cerebellar ataxic phenotype in SCA1. In this study, we report progressive dysfunction of metabotropic glutamate receptor (mGluR) signalling, which leads to smaller slow synaptic responses, reduced dendritic Ca2+ signals and impaired synaptic plasticity at cerebellar synapses, in the early disease stage of SCA1 model mice. We also show that enhancement of mGluR signalling by a clinically available drug, baclofen, leads to improvement of motor performance in SCA1 mice. SCA1 is an incurable disease with no effective treatment, and our results may provide mechanistic grounds for targeting mGluRs and a novel drug therapy with baclofen to treat SCA1 patients in the future.
Abstract: Spinocerebellar ataxia type 1 (SCA1) is a progressive neurodegenerative disease that presents with cerebellar ataxia and motor learning defects. Previous studies have indicated that the pathology of SCA1, as well as other ataxic diseases, is related to signalling pathways mediated by the metabotropic glutamate receptor type 1 (mGluR1), which is indispensable for proper motor coordination and learning. However, the functional contribution of mGluR signalling to SCA1 pathology is unclear. In the present study, we show that SCA1 model mice develop a functional impairment of mGluR signalling which mediates slow synaptic responses, dendritic Ca2+ signals, and short- and long-term synaptic plasticity at parallel fibre (PF)-Purkinje cell (PC) synapses in a progressive manner from the early disease stage (5 postnatal weeks) prior to PC death. Notably, impairment of mGluR-mediated dendritic Ca2+ signals linearly correlated with a reduction of PC capacitance (cell surface area) in disease progression. Enhancement of mGluR signalling by baclofen, a clinically available GABAB receptor agonist, led to an improvement of motor performance in SCA1 mice and the improvement lasted ∼1 week after a single application of baclofen. Moreover, the restoration of motor performance in baclofen-treated SCA1 mice matched the functional recovery of mGluR-mediated slow synaptic currents and mGluR-dependent short- and long-term synaptic plasticity. These results suggest that impairment of synaptic mGluR cascades is one of the important contributing factors to cerebellar ataxia in early and middle stages of SCA1 pathology, and that modulation of mGluR signalling by baclofen or other clinical interventions may be therapeutic targets to treat SCA1.
Keywords: calcium imaging; cerebellar ataxia; metabotropic glutamate receptor; purkinje cell; synaptic plasticity.
© 2016 The Authors. The Journal of Physiology © 2016 The Physiological Society.
Figures
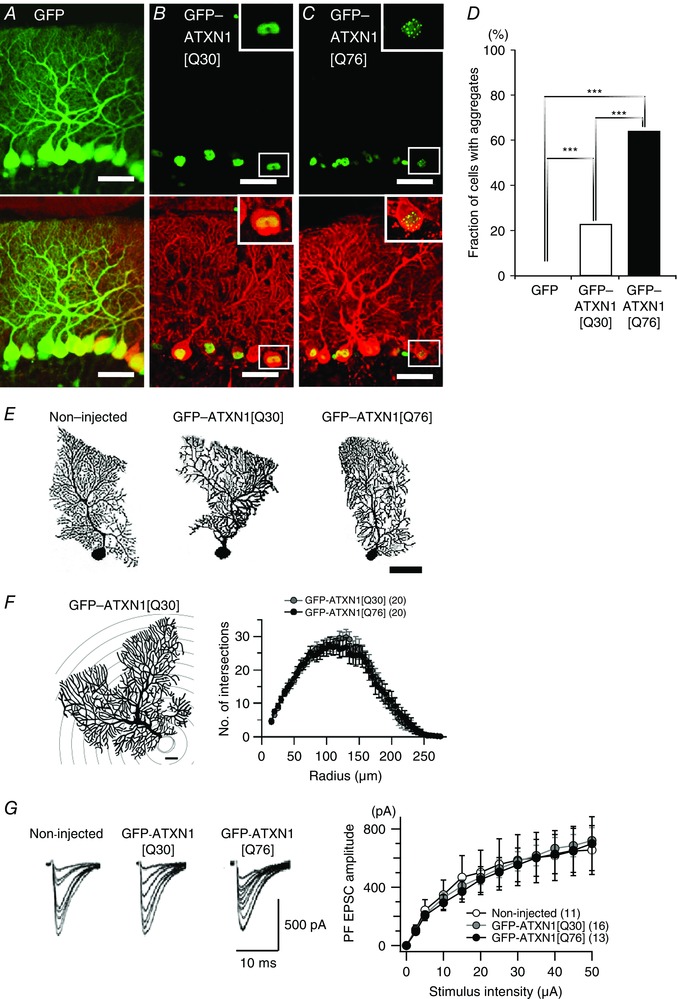
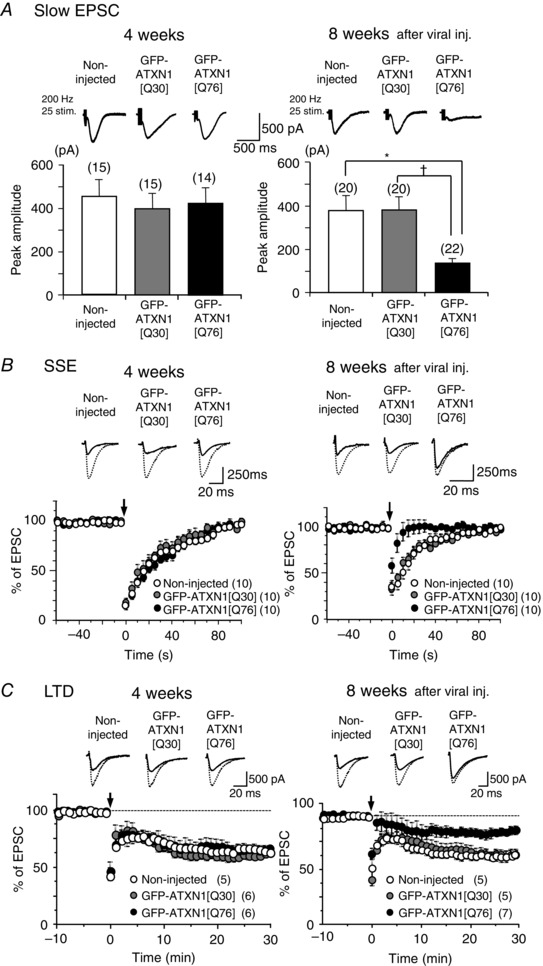
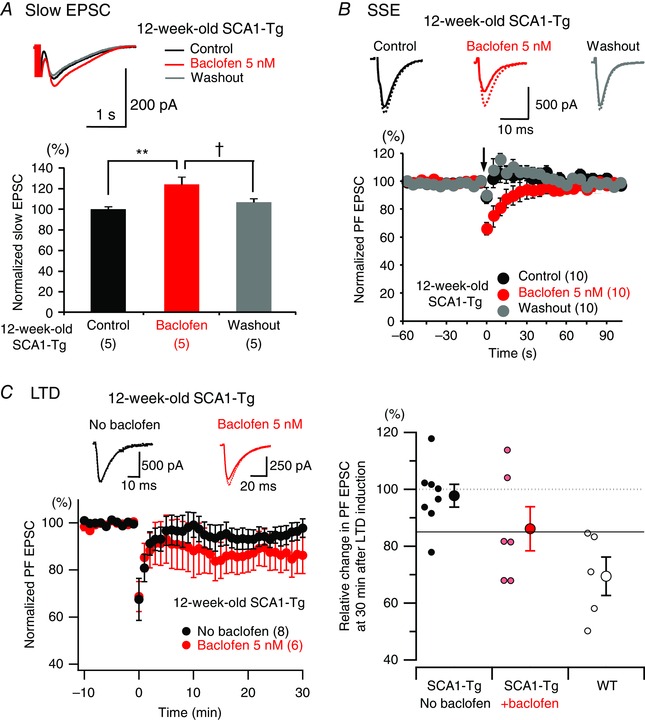
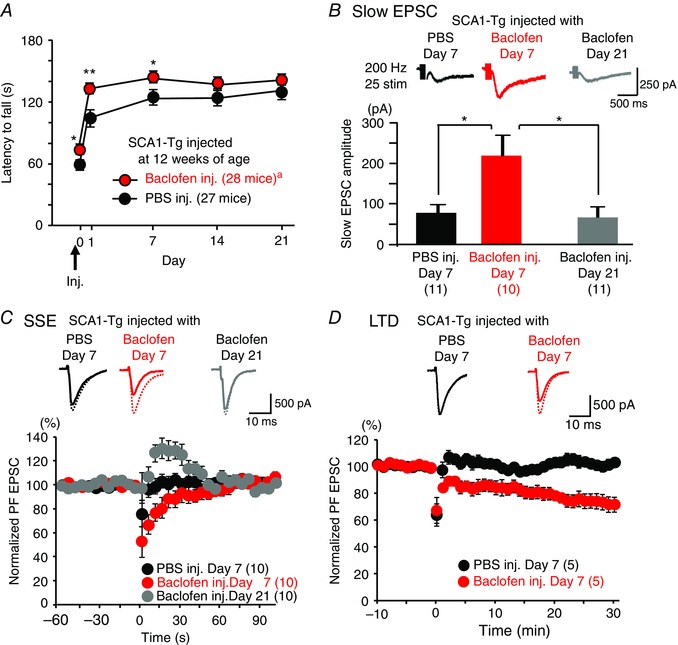
Comment in
-
New old drug(s) for spinocerebellar ataxias.J Physiol. 2017 Jan 1;595(1):5-6. doi: 10.1113/JP273149. J Physiol. 2017. PMID: 28035676 Free PMC article. No abstract available.
References
-
- Aiba A, Kano M, Chen C, Stanton ME, Fox GD, Herrup K, Zwingman TA & Tonegawa S (1994). Deficient cerebellar long‐term depression and impaired motor learning in mGluR1 mutant mice. Cell 79, 377–388. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous