

Genetic analysis of Tunisian families with Usher syndrome type 1: toward improving early molecular diagnosis
- PMID: 27440999
- PMCID: PMC4950652
Genetic analysis of Tunisian families with Usher syndrome type 1: toward improving early molecular diagnosis
Abstract
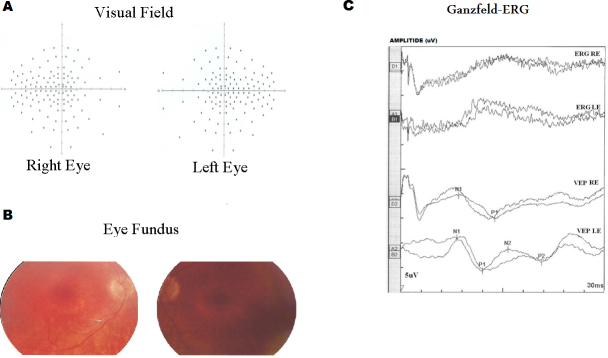
Purpose: Usher syndrome accounts for about 50% of all hereditary deaf-blindness cases. The most severe form of this syndrome, Usher syndrome type I (USH1), is characterized by profound congenital sensorineural deafness, vestibular dysfunction, and retinitis pigmentosa. Six USH1 genes have been identified, MYO7A, CDH23, PCDH15, USH1C, SANS, and CIB2, encoding myosin VIIA, cadherin-23, protocadherin-15, harmonin, scaffold protein containing ankyrin repeats and a sterile alpha motif (SAM) domain, and calcium- and integrin-binding member 2, respectively.
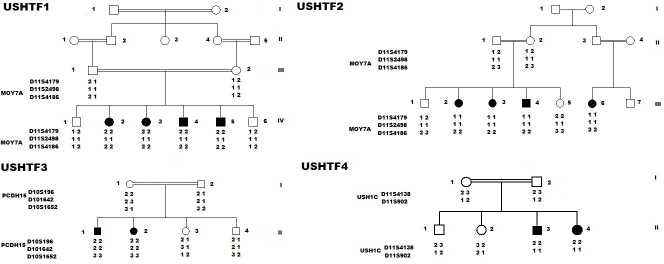
Methods: In the present study, we recruited four Tunisian families with a diagnosis of USH1, together with healthy unrelated controls. Affected members underwent detailed audiologic and ocular examinations. We used the North African Deafness (NADf) chip to search for known North African mutations associated with USH. Then, we selected microsatellite markers covering USH1 known loci to genotype the DNA samples. Finally, we performed DNA sequencing of three known USH1 genes: MYO7A, PCDH15, and USH1C.
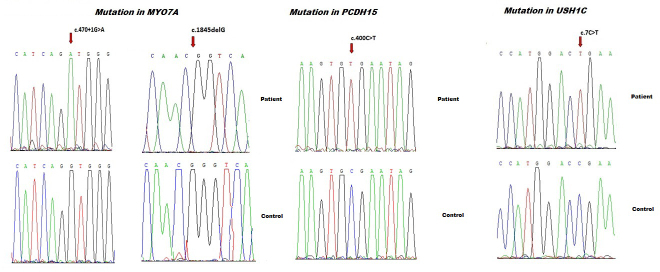
Results: Four biallelic mutations, all single base changes, were found in the MYO7A, USH1C, and PCDH15 genes. These mutations consist of a previously reported splicing defect c.470+1G>A in MYO7A, three novel variants, including two nonsense (p.Arg3X and p.Arg134X) in USH1C and PCDH15, respectively, and one frameshift (p.Lys615Asnfs*6) in MYO7A.
Conclusions: We found a remarkable genetic heterogeneity in the studied families with USH1 with a variety of mutations, among which three were novel. These novel mutations will be included in the NADf mutation screening chip that will allow a higher diagnosis efficiency of this extremely genetically heterogeneous disease. Ultimately, efficient molecular diagnosis of USH in a patient's early childhood is of utmost importance, allowing better educational and therapeutic management.
Figures
References
-
- Keats BJ, Corey DP. The Usher syndromes. Am J Med Genet. 1999;89:158–66. - PubMed
-
- Bonnet C, El-Amraoui A. Usher syndrome (sensorineural deafness and retinitis pigmentosa): Pathogenesis, molecular diagnosis and therapeutic approaches. Curr Opin Neurol. 2012;25:42–9. - PubMed
-
- Smith RJ, Berlin CI, Hejtmancik JF, Keats BJ, Kimberling WJ, Lewis RA, Moller CG, Pelias MZ, Tranebjaerg L. Clinical diagnosis of the Usher syndromes. Usher Syndrome Consortium. Am J Med Genet. 1994;50:32–8. - PubMed
-
- Kinga MB, Mark C, Emily P, Shyana H, Jaclyn L, Daniel GT, Joseph W, Daniel NG, Carol WD, Michael HF, Xiaowu G, Eliot LB. Targeted Exon Sequencing in Usher Syndrome Type I. Biochemistry and Molecular Biology. 2014;55:8488–96. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
