

Host-Pathogen Coevolution and the Emergence of Broadly Neutralizing Antibodies in Chronic Infections
- PMID: 27442127
- PMCID: PMC4956326
- DOI: 10.1371/journal.pgen.1006171
Host-Pathogen Coevolution and the Emergence of Broadly Neutralizing Antibodies in Chronic Infections
Abstract
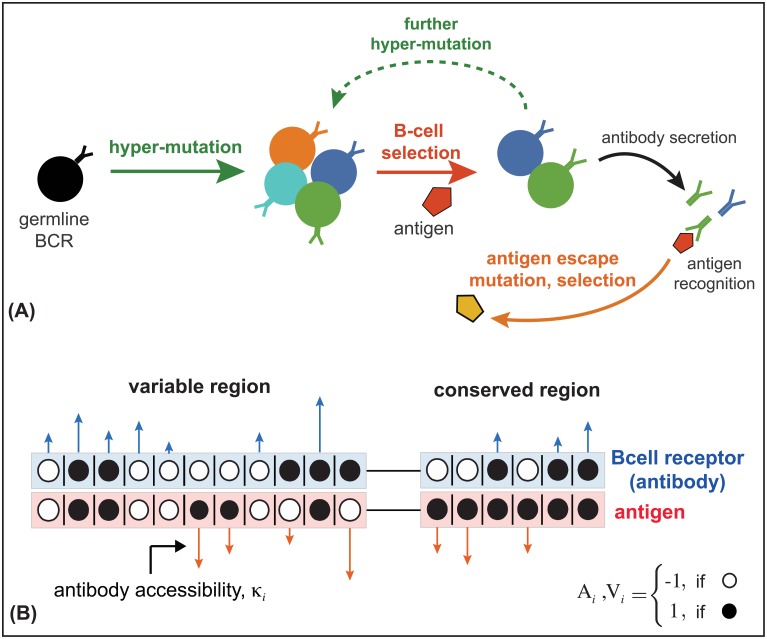
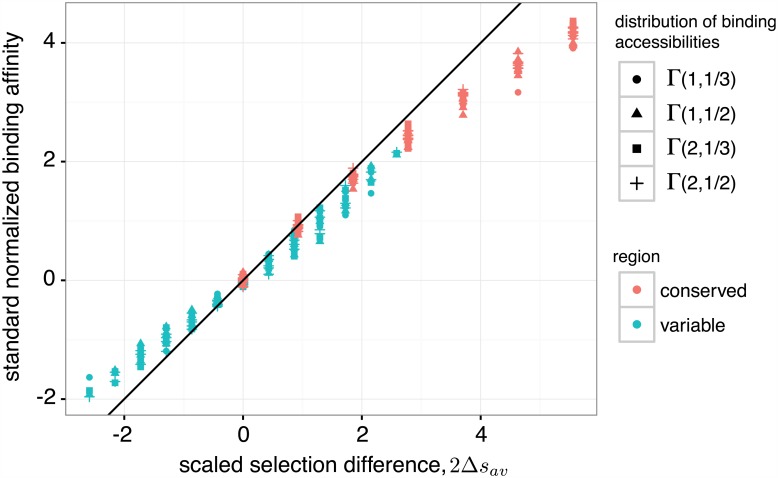
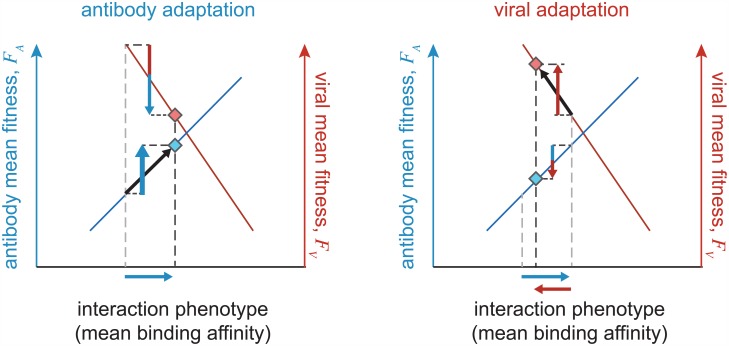
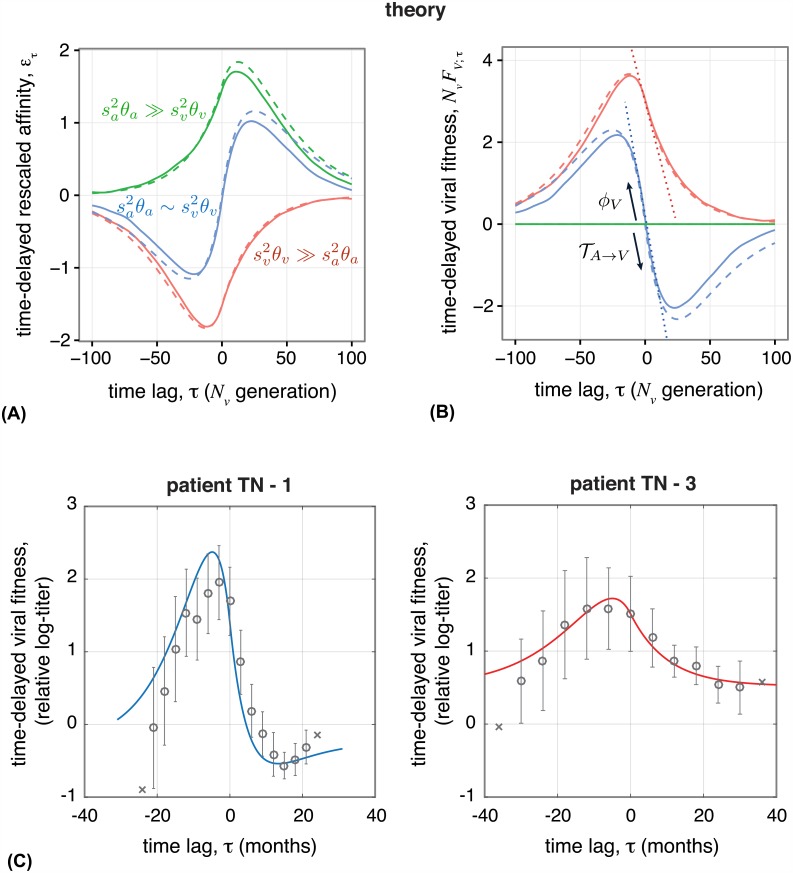
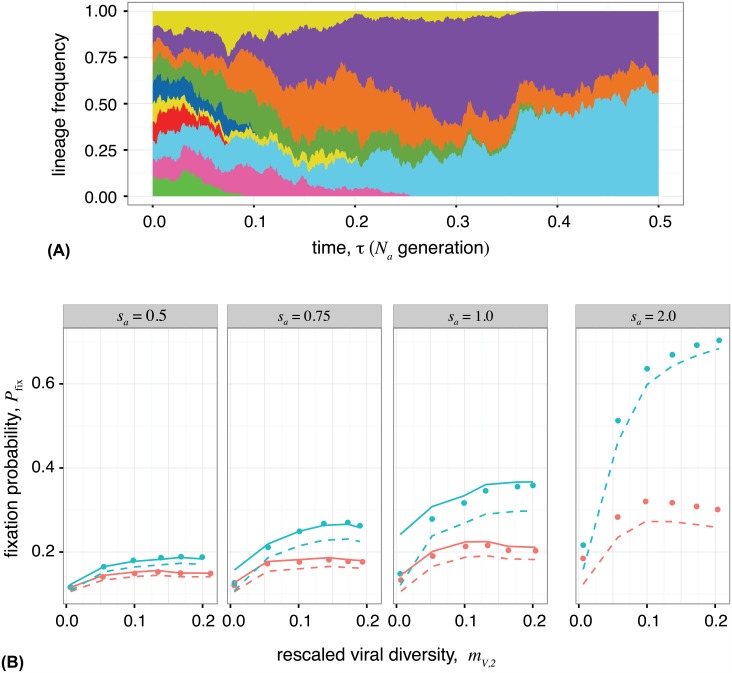
The vertebrate adaptive immune system provides a flexible and diverse set of molecules to neutralize pathogens. Yet, viruses such as HIV can cause chronic infections by evolving as quickly as the adaptive immune system, forming an evolutionary arms race. Here we introduce a mathematical framework to study the coevolutionary dynamics between antibodies and antigens within a host. We focus on changes in the binding interactions between the antibody and antigen populations, which result from the underlying stochastic evolution of genotype frequencies driven by mutation, selection, and drift. We identify the critical viral and immune parameters that determine the distribution of antibody-antigen binding affinities. We also identify definitive signatures of coevolution that measure the reciprocal response between antibodies and viruses, and we introduce experimentally measurable quantities that quantify the extent of adaptation during continual coevolution of the two opposing populations. Using this analytical framework, we infer rates of viral and immune adaptation based on time-shifted neutralization assays in two HIV-infected patients. Finally, we analyze competition between clonal lineages of antibodies and characterize the fate of a given lineage in terms of the state of the antibody and viral populations. In particular, we derive the conditions that favor the emergence of broadly neutralizing antibodies, which may have relevance to vaccine design against HIV.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Janeway C. A, Travers P, Walport M & Shlomchik M. (2005) Immunobiology: The Immune System in Health and Disease (Garland Science, New York: ).
-
- Glanville J, Zhai W, Berka J, Telman D, Huerta G, Mehta G. R & et al. (2009) Precise determination of the diversity of a combinatorial antibody library gives insight into the human immunoglobulin repertoire. Proc. Natl. Acad. Sci. U.S.A. 106, 20216–20221. 10.1073/pnas.0909775106 - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
